
Abstract
Amyloidosis is a group of diseases in which proteins become amyloid, an insoluble fibrillar aggregate, resulting in organ dysfunction. Amyloid deposition has been reported in various animal species. To diagnose and understand the pathogenesis of amyloidosis, it is important to identify the amyloid precursor protein involved in each disease. Although 42 amyloid precursor proteins have been reported in humans, little is known about amyloidosis in animals, except for a few well-described amyloid proteins, including amyloid A (AA), amyloid light chain (AL), amyloid β (Aβ), and islet amyloid polypeptide-derived amyloid. Recently, several types of novel amyloidosis have been identified in animals using immunohistochemistry and mass spectrometry–based proteomic analysis. Certain species are predisposed to specific types of amyloidosis, suggesting a genetic background for its pathogenesis. Age-related amyloidosis has also emerged due to the increased longevity of captive animals. In addition, experimental studies have shown that some amyloids may be transmissible. Accurate diagnosis and understanding of animal amyloidosis are necessary for appropriate therapeutic intervention and comparative pathological studies. This review provides an updated classification of animal amyloidosis, including associated protein misfolding disorders of the central nervous system, and the current understanding of their pathogenesis. Pathologic features are presented together with state-of-the-art diagnostic methods that can be applied for routine diagnosis and identification of novel amyloid proteins in animals.
Keywords
Amyloidosis is a group of diseases characterized by the deposition of insoluble, fibrillar protein aggregates, termed amyloid, in single or multiple organs, resulting in organ dysfunction. 24 Since amyloid is an insoluble fibrillar aggregate derived from native precursor proteins, amyloidosis is classified by its precursor protein. 24 According to the International Society of Amyloidosis, 42 amyloid precursor proteins have been reported in humans to date. 24
Systemic amyloidoses had been further classified into “primary amyloidosis” and “secondary (reactive) amyloidosis,” corresponding to amyloid light chain (AL) amyloidosis and amyloid A (AA) amyloidosis, respectively.103,168,241 However, since AA amyloidosis is not always accompanied by a well-defined underlying inflammatory lesion and can be a primary disease, 229 these two classifications should no longer be used. In recent years, it has been revealed that a wide variety of proteins may form amyloid, in addition to well-known amyloid precursor proteins such as serum amyloid A (SAA),137,168 immunoglobulin light chain,80,138,204 the amyloid β precursor protein (APP),243,246 and islet amyloid polypeptide (IAPP).109,184 Although AA amyloidosis is the most common type of systemic amyloidosis, recent studies have shown that other forms of amyloidosis, such as apolipoprotein A-IV (ApoA-IV) amyloidosis and fibrinogen Aα-chain amyloidosis, also present as systemic amyloidosis in animals.97,224
Amyloid typing is necessary to determine the etiology and treatment strategy for each amyloidosis. For example, AA amyloidosis and AL amyloidosis are associated with chronic inflammation and plasmacytic proliferative disease, respectively.5,166 In addition, some amyloidoses are associated with other neoplastic diseases148,169,170 or aging.84,97
This review provides an updated classification of animal amyloidoses (Table 1) and the current understanding of their pathogeneses. Pathological features are presented together with their etiology and available diagnostic methods for routine diagnosis and future identification of novel amyloid proteins.
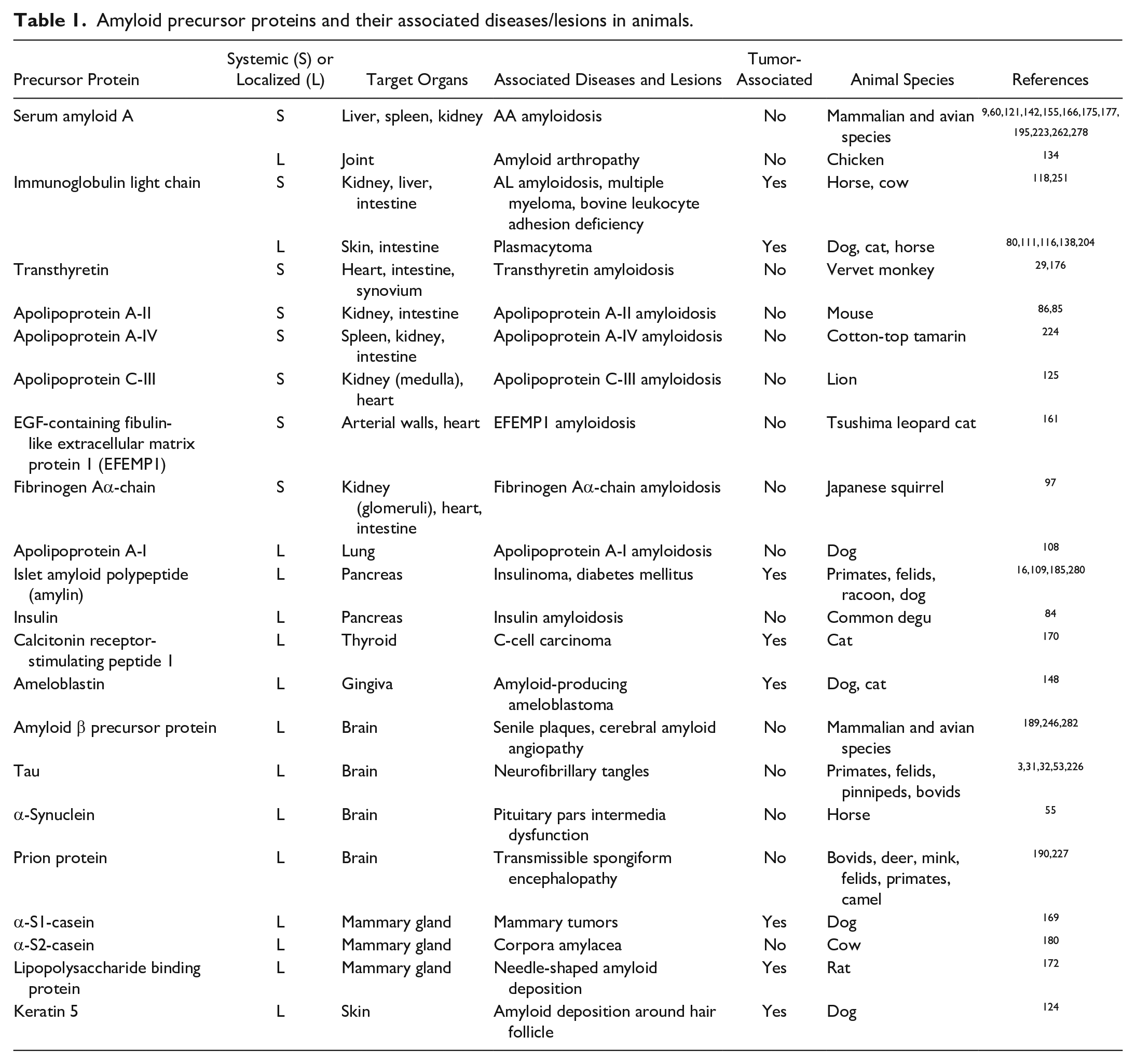
Amyloid precursor proteins and their associated diseases/lesions in animals.
Terminology
Amyloid
Amyloid is an insoluble fibrillar protein with a cross-β structure resulting from misfolding of native amyloid precursor protein. 24 The basic unit of an amyloid fibril is a protofilament, a stack of monomers forming a β-sheet, which twists with other stacks to form the fibril. 24 Amyloid fibrils are highly ordered and have stiffer properties than biological filaments such as actin. 123 Amyloid fibrils possess greater stability and resistance to degradation by chemical or biological means than the physiological state of the precursor proteins. 82
Amyloid was classically defined as extracellularly formed proteinous materials. 274 Thereafter, aggregates of tau and α-synuclein were reported in association with various neurodegenerative diseases.206,252 Since these aggregates are negative for Congo red staining (a gold standard method for amyloid identification, described below) and form intracellularly in neurons and glial cells, they have not been considered as amyloid but as “misfolded proteins” like prion. 94 However, these aggregates can also contain fibrils with a cross-β structure, share a common pathological nature with “classical amyloid,” characterized by cellular degeneration and proteostatic disorders, and can sometimes be found extracellularly. Therefore, the term amyloidosis in a broader sense, including such misfolding diseases, reflects current usage. 230 In view of updated trends in nomenclature in the medical field, 24 this review also defines amyloid as a fibrillar protein with a cross-β structure. In other words, this review agrees to use the term amyloid not only for classical Congo red–positive pathological deposits but also for intracellular aggregates, functional amyloid, and in vitro synthetic fibrils, if they meet this definition. On the other hand, it is currently technically challenging to accurately characterize the nature of intracellular aggregates. For example, although tau and α-synuclein have demonstrated that some aggregates have amyloid properties, the pathology of tauopathy and synucleinopathy is not uniform, and it is not clear whether amyloid or nonamyloid aggregates are associated with the pathogenesis of these diseases.66,91,154 With these understandings in mind, and although there are still ongoing debates, tau, α-synuclein, and prions are discussed as amyloid in this review.
Amyloidosis
Amyloidosis is a disease characterized by amyloid deposition in various tissues that results in functional abnormalities. 24 A small amount of amyloid deposits with no pathogenicity may be found incidentally on histologic examination without showing any related clinical signs. In such cases, it is appropriate to describe the histopathological findings (i.e., amyloid deposits) rather than diagnose amyloidosis (discussed below). In addition, although some neurodegenerative conditions present with amyloid formation, it may not be known whether amyloid or nonamyloid aggregates are involved in the pathogenesis. For these diseases, the term “protein misfolding disorder” seems to be more appropriate than “amyloidosis.”
Amyloid Signature Protein
Amyloid signature proteins are proteins that are not amyloid but colocalize with various amyloid proteins and are thought to be involved in amyloid deposition or inhibition. 24 Various amyloid signature proteins have been identified in humans and animals, including apolipoprotein E (ApoE), ApoA-IV, and clusterin.24,159,160 The profiles of amyloid signature proteins differ among amyloid precursor proteins, affected tissues, and animal species (Table 2). The serum amyloid P-component (SAP) is an uncommon amyloid signature protein in animal amyloidosis, although it is frequently detected in human amyloidosis.72,193 In fact, SAP colocalizes with primate amyloid16,92,100 but is not detected in canine or feline amyloidosis.59,160 Furthermore, SAP colocalizes with AA in adult pigs, but not in young pigs. 96 Previous studies of amyloidosis have focused mainly on amyloidogenic proteins, and the nature of amyloid signature proteins is still largely unknown. However, it is not difficult to imagine that these proteins, which colocalize or bind to amyloid fibrils, play an important role in the pathogenesis of amyloidosis.
Differences in amyloid signature proteins by amyloid precursor proteins and animal species.
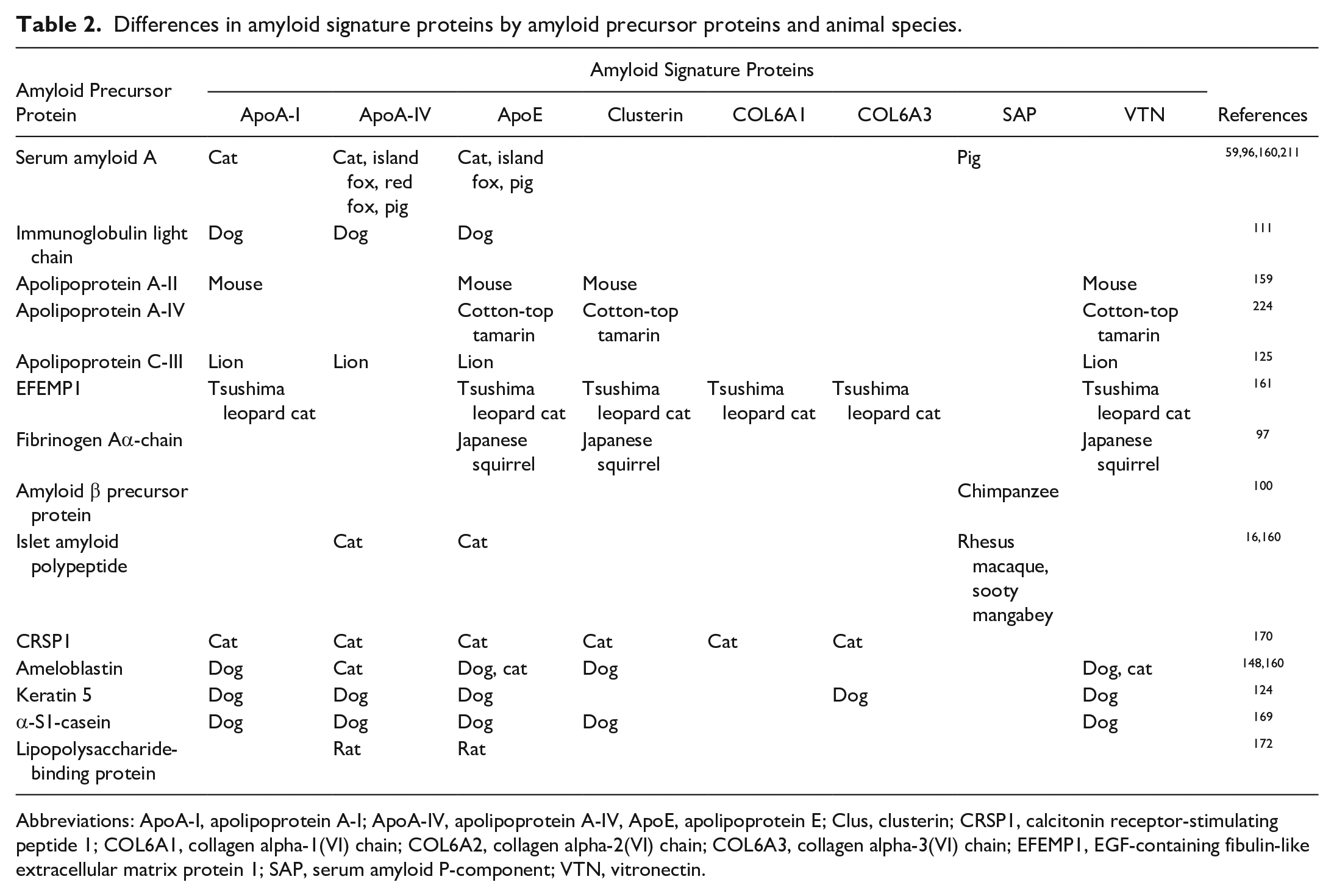
Abbreviations: ApoA-I, apolipoprotein A-I; ApoA-IV, apolipoprotein A-IV, ApoE, apolipoprotein E; Clus, clusterin; CRSP1, calcitonin receptor-stimulating peptide 1; COL6A1, collagen alpha-1(VI) chain; COL6A2, collagen alpha-2(VI) chain; COL6A3, collagen alpha-3(VI) chain; EFEMP1, EGF-containing fibulin-like extracellular matrix protein 1; SAP, serum amyloid P-component; VTN, vitronectin.
Amyloid Formation
Although the factors contributing to amyloid formation in vivo are diverse and complex, nucleation-dependent polymerization is considered the universal principle of amyloid formation (Fig. 1).174,212 In the initial step of amyloidosis, a small number of precursor proteins are converted to intermediate species (specifically oligomers), which in turn can act as nuclei and facilitate further polymerization into protofibrils and fibrils by gathering precursor proteins. The initial small amyloid aggregate is often described as a “seed” because of its potential to grow into large aggregates and may even propagate to other organs in the body and form large aggregates or new seeds. Factors that initiate amyloidogenesis include (1) changes in stability or aggregability due to amino acid mutations or posttranslational modifications,63,77,201 (2) destabilization of multimeric structures by mutations, 144 (3) increased self-interaction due to supersaturation of precursor proteins,71,127,269 and (4) destabilization by proteolysis of full-length proteins.169,212,236 In addition, other environmental factors, such as oxidative stress and free radicals, have been implicated in protein misfolding. 1 The speed of amyloid aggregation largely depends on the concentration of the amyloid precursor protein in the microenvironment surrounding seeds. 212
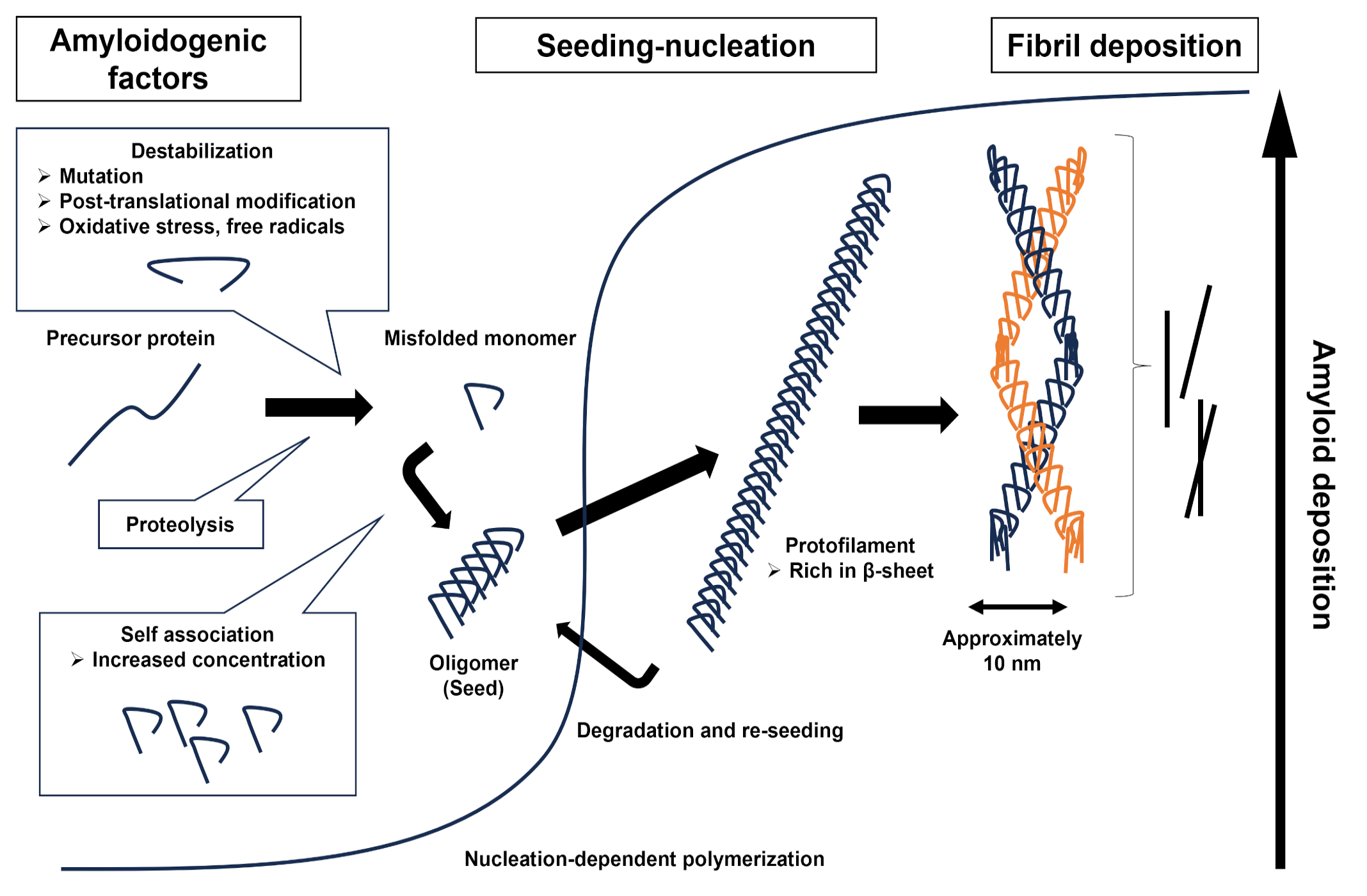
Diagram of amyloid fibril formation. Many factors, such as mutations, proteolysis, and self-association of amyloid precursor proteins, may enhance misfolding and oligomerization. Oligomers can act as seeds that accelerate amyloid fibril formation in tissues. Elongated protofibrils twist together and form amyloid fibrils that are approximately 10 nm in diameter.
As described later, prions are transmissible among individuals, although they are not organisms with their own genome like virus or bacteria. 4 Prion-like transmission and seeding properties have been experimentally demonstrated for other pathogenic amyloidogenic proteins such as SAA, apolipoprotein A-II (ApoA-II), tau, and α-synuclein.4,131,149,168 Some researchers have suggested collectively calling such amyloid-related diseases “prion disorders.”8,26
Diagnostic Methods for Amyloidosis
Histologic Diagnosis
The identification of amyloidosis begins with the suspicion of amyloid deposits on hematoxylin and eosin–stained sections during routine diagnostic histologic evaluation, which can then be confirmed by additional special staining, particularly Congo red staining. Immunohistochemistry or mass spectrometry alone, described below, cannot confirm the diagnosis. 224 Typically, amyloids in hematoxylin and eosin–stained sections can be observed as amorphous eosinophilic or pale basophilic material in the stroma and vascular walls. 130 For some types of amyloid, the precursor protein can be predicted based on their deposition patterns. The histologic characteristics of each amyloid are described below.
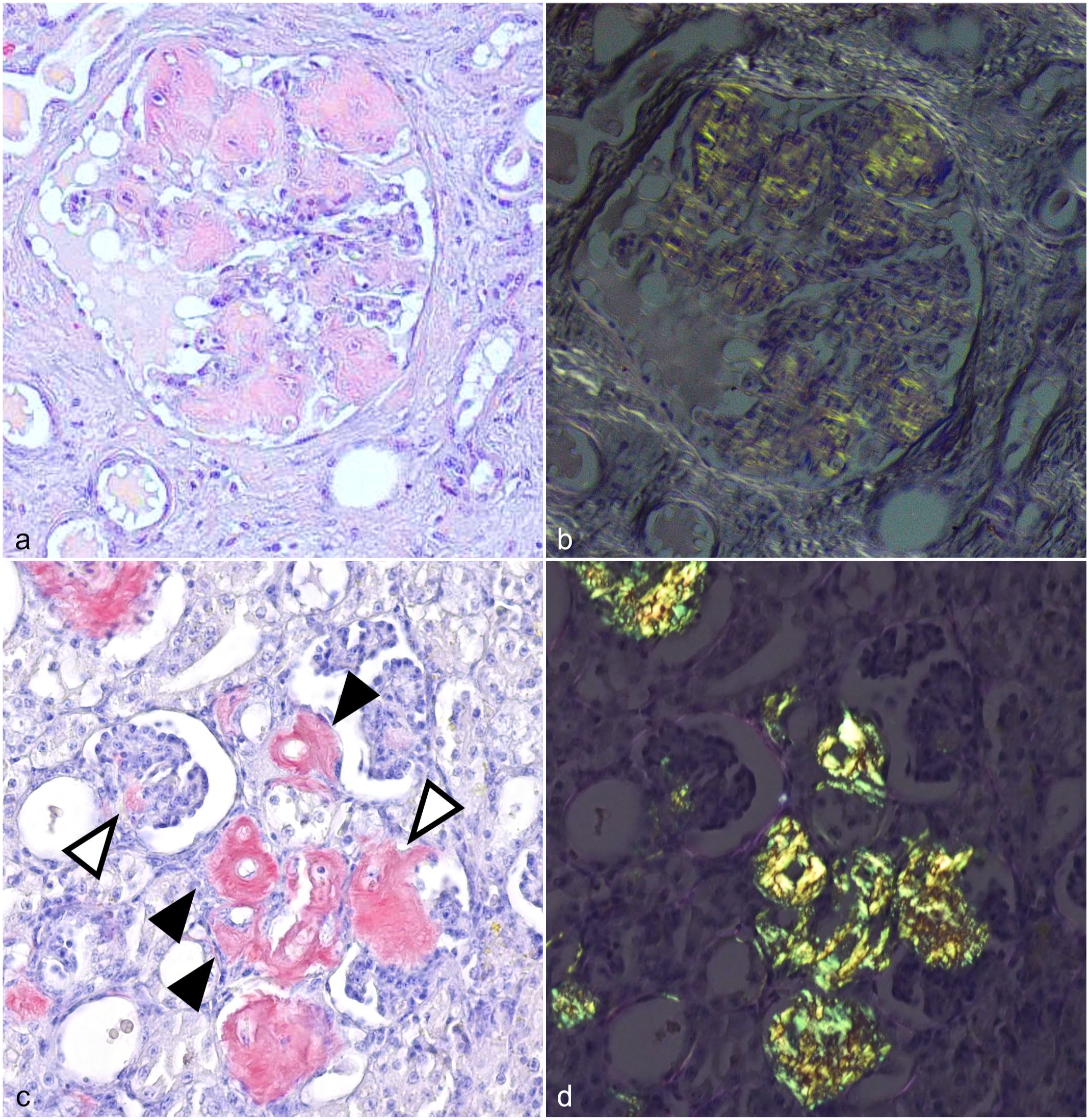
Congo red is commonly used for the histologic detection of amyloid. 277 Although several methods for Congo red staining have been proposed, the authors recommend the alkaline Congo red staining method, which is currently widely used. 277 Congo red–stained amyloid deposits show orange to red color in bright field (Fig. 2a, c), yellow to green birefringence under polarized light (Fig. 2b, d), and orange fluorescence under blue to green excitation light. 277 The regular fitting of Congo red molecules into the molecular pockets on the surface of amyloid fibrils creates a crystal-like regularly ordered Congo red orientation, which may contribute to dichroic birefringence. 277 Collagen and elastic fibers can also be stained with Congo red nonspecifically 277 but can be distinguished from amyloid by their white birefringence under polarized light (Fig. 2b). Staining intensity may vary according to the severity of amyloid deposition, background tissue, animal species, and type of amyloid (Fig. 2). Notably, some amyloids, such as α-synuclein and tau, do not or poorly stain with Congo red. 218
Congo red staining of AA amyloid in different animal species.
The potassium permanganate method has been used for amyloid typing. In this method, the yellow to green birefringence by Congo red staining is attenuated after potassium permanganate treatment in AA, whereas the staining is retained in AL. 29 However, AA in some animal species is resistant to potassium permanganate treatment, and potassium permanganate–sensitive AL has also been reported. 111 Therefore, its use is considered unreliable.
Thioflavin S, a fluorescent dye, has also been used as a specific labeling method for amyloid fibrils.10,98 Thioflavin T is commonly used for quantification of amyloid fibrils in vitro, using a fluorescence spectrophotometer. 15 It can also be used for amyloid staining on tissue specimens using a fluorescent microscope. 100
Transmission Electron Microscopy
Amyloid fibrils are ultrastructurally defined as unbranched fibrils approximately 10 nm in diameter.137,161,277 Some amyloids that are negative for Congo red can be confirmed by electron microscopy. 217
Immunohistochemistry
Immunohistochemistry is a useful tool for determining amyloid precursor proteins, but it is necessary to validate antibody cross-reactivity when applied to proteins of different animal species. 203 It should be noted that inter- and intra-species variations (polymorphisms) in the amino acid sequence of the target antigen may be associated with the amyloidogenicity of the protein. Protein databases, such as the National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/), are useful for comparing amino acid sequences between species and estimating the antigenic affinity to the antibody and amyloidogenicity of the protein.
In some amyloids, it may be difficult for antibodies to bind to the epitope because of the conformational changes in the native structure. In such cases, denaturation with formic acid is effective for antigen retrieval. False-negative results may be obtained if the immunoreaction-targeted amyloidogenic regions are poorly conserved.124,148 In such cases, it is advisable to use multiple antibodies targeting different regions of the protein, including nonamyloidogenic regions, which can be predicted by using a structural prediction software program.70,265 For instance, AL amyloid fibrils are composed of an immunoglobulin variable domain, and thus, antibody targeting the C-terminal constant domain yields better immunohistochemical results.200,239
Positive immunolabeling does not necessarily indicate that the target antigen is an amyloid. As mentioned earlier, many amyloid signature proteins colocalize with amyloids, and it is important not to misinterpret amyloid signature proteins as amyloids. Also, nonamyloid native amyloid precursor proteins may be detected by immunohistochemistry. Because amyloid is resistant to proteolysis, enzymatic treatment with actinase or proteinase K prior to primary antibody sensitization may differentiate amyloid from its precursor protein.172,188,268
Antibodies for detecting specific protein conformational changes or conformers, such as oligomers and fibrils, are commercially available for some proteins. 27 Antibodies for detecting proteins with posttranscriptional modifications, such as phosphorylation, are also available. 27 Western blotting should be performed for antibody validation to confirm antigenicity and specificity for conformational posttranscriptional changes.
Mass Spectrometry
Mass spectrometry comprehensively analyzes peptides and protein constituents based on their molecular weights and charges. 141 However, it relies on currently available protein databases. Consequently, animal proteins not registered in the database, mostly of nondomestic species, or whose exact amino acid sequence differs from that of the registered database because of variations (polymorphisms), are often difficult to identify. In such cases, de novo sequencing validation is necessary. 97 Also, analytical techniques should consider the various potential modifications of proteins.36,122
For mass spectrometry–based amyloid analysis, it is important that the sample is selectively collected from amyloid deposits in the tissue specimens. Laser microdissection is used to precisely collect samples from the region of interest on a tissue section. Laser microdissection followed by liquid chromatography-tandem mass spectrometry provides a list of peptides that comprise the proteins in the deposits. 141 This technique is useful for amyloid typing in humans and animals.112,124,161,178,211 If the area of the amyloid deposit is sufficiently large (>100,000 µm2), it can be manually dissected under a stereomicroscope.148,170,224 The advantage of manual dissection is that amyloid can be collected from regular glass slides, and no expensive equipment is necessary. Proteomic analysis can also be performed using archived, stained tissue slides. 170 As mentioned above, amyloid signature proteins such as ApoE and ApoA-IV are co-deposited with amyloid fibrils. The detection of multiple amyloid signature proteins by mass spectrometry can validate the accuracy of tissue dissection.
Classification of Amyloidosis
To date, 21 amyloid precursor proteins have been identified in animals (Table 1). Among these, ameloblastin, α-S1-casein, α-S2-casein, calcitonin receptor-stimulating peptide 1 (CRSP1), and lipopolysaccharide-binding protein have been reported only in animals, and others have also been reported in humans. 24 Some amyloid deposition syndromes are of unknown clinical significance, as discussed below.
Amyloid deposition can occur in multiple organs when the precursor is a serum protein, resulting in systemic amyloidosis. Amyloid deposition confined to a specific organ or lesion area (e.g. tumor) is referred to as localized amyloidosis. Some amyloids, namely AA and AL, may present as either systemic or localized amyloidosis. Most localized amyloidoses are associated with neoplastic diseases and/or aging. However, the clinical significance of amyloid deposition in some forms of localized amyloidosis remains unclear.
Serum AA
Animals express up to four SAA isoforms, SAA1 through SAA4, of which SAA1 is involved in AA amyloidosis.122,168,242 AA amyloidosis is the most common systemic amyloidosis in animals and has been reported in various mammalian9,60,112,121,137,142,155,175,195,223,262,278 and avian species.35,166,177,178,189 As an exception, rats do not develop AA amyloidosis because they do not express full-length SAA. 153 AA amyloidosis is commonly a systemic disease, but localized conditions such as amyloid arthropathy in chickens have been reported.121,122
The precursor protein SAA is an acute-phase protein synthesized in the liver in response to cytokines such as interleukin-6. 214 Amyloid fibril formation is commonly triggered by supersaturation of circulating SAA associated with persistent inflammation. 269 However, cases without apparent underlying diseases or familial conditions due to SAA mutations have also been reported. Familial AA amyloidosis has been reported in some canine and feline breeds, including Shar-Pei, 225 Bracco Italiano, 95 Siamese, and Abyssinians.20,262
Amyloid deposits can be found in vessel walls and interstitium throughout the body. In the kidney, the most commonly affected organ, severe amyloid deposition in glomeruli causes nephrotic syndrome (Fig. 3a). Diffuse deposition in the perisinusoidal space and the vascular walls is commonly observed in the liver (Fig. 3b). In the spleen, diffuse deposition in the perifollicular and perivascular areas can be seen (Fig. 3c). The lamina propria is the predominant location of amyloid deposition in the gastrointestinal tract (Fig. 3d). In addition, amyloid deposition can be observed in the vascular walls and interstitium of the pancreas, reproductive organs, endocrine organs (Fig. 3e), and other tissues (Fig. 3f). Although AA deposition in the nervous system is extremely rare, peripheral nervous system involvement has been reported in flamingos (Fig. 3g). 189
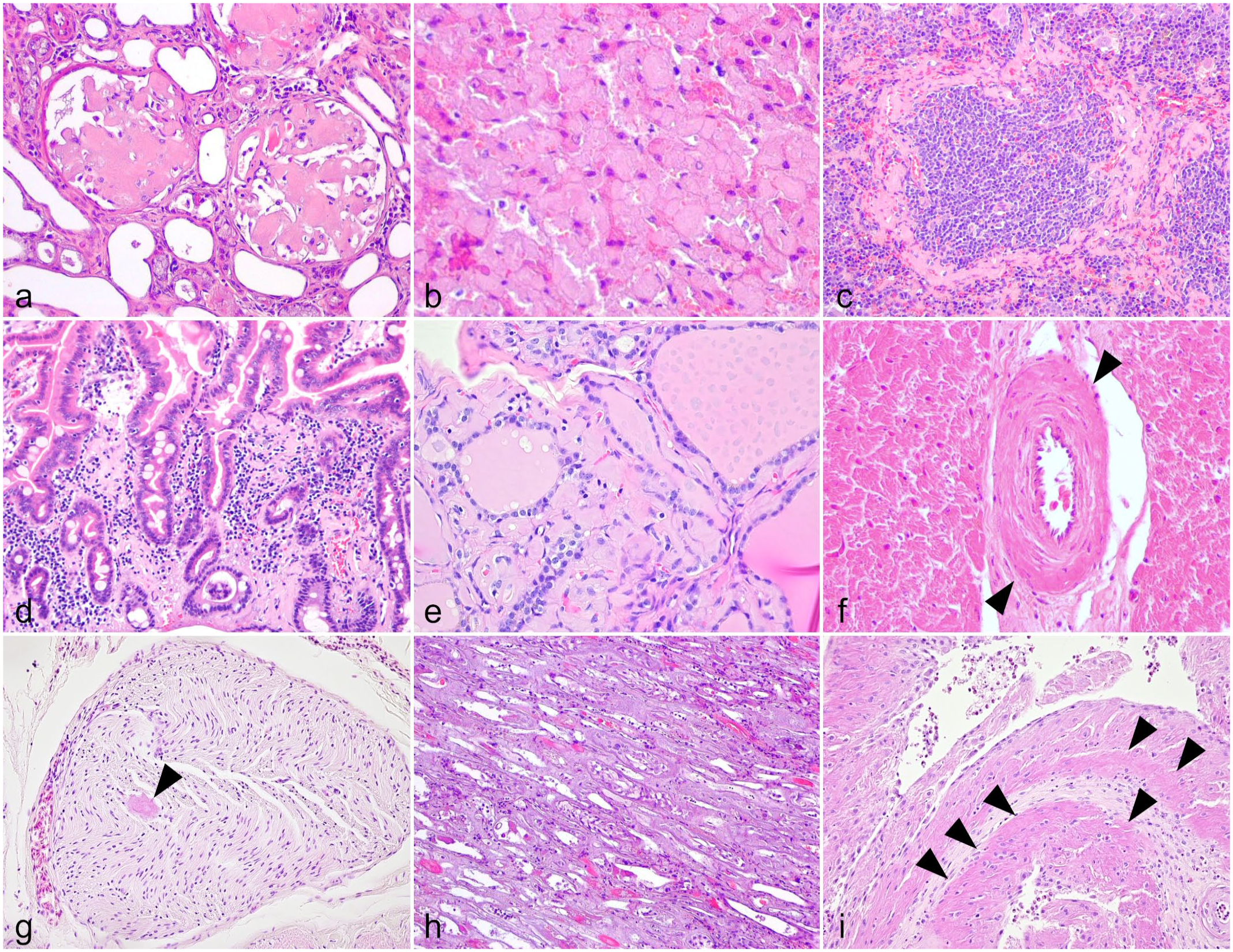
Histologic features of AA amyloidosis in animals. Hematoxylin and eosin.
Deposition patterns of AA can vary among animal species and individual cases. For instance, glomerular deposition is rare, and medullary deposition is predominant in felids (Fig. 3h).195,223 Atypical cases in cows have been reported in which renal deposition is almost absent, and deposition is predominant in the liver. 165 In experimentally induced murine AA amyloidosis models, amyloid deposition is relatively rare in the glomeruli and is often more prominent in the renal medulla.99,267
AA amyloidosis is the primary differential diagnosis for systemic amyloidosis in animals. Immunohistochemistry using anti-SAA antibodies confirms the diagnosis. Commercially available anti-human SAA monoclonal antibodies (clones mc1 and 115) have very low cross-reactivity with animal AA. The polyclonal anti-human SAA antibody PAA885Hu01 (Cloud-Clone Corp., Houston, TX) is useful for detecting AA in animals. There is a highly conserved region in the SAA protein among various animal species 58 ; thus, antibody targeting this sequence is recommended. 99
Transmission of AA amyloidosis has been demonstrated in various experimental animals.143,162,167,171 Intravenously, intraperitoneally, or even orally administered exogenous AA is thought to act as a seed in the recipient and promote nucleation-dependent polymerization of the host SAA. 143 Transmission of AA amyloidosis has been shown to occur between different species.39,231 In addition, experimental studies have shown that AA can be transmitted by oral ingestion. 168 A high incidence of AA amyloidosis has been reported in flocks of chickens, 171 cheetahs, 283 and sheltered cats, 51 suggesting that horizontal transmission may occur between animals. Recent detailed structural analysis of amyloid fibrils using cryo-electron microscopy has shown that the N-terminal structures of human and murine AA fibrils are very similar, which may contribute to the stability of the amyloid fibrils and seeding. 136 Detailed risk analyses are necessary to evaluate the potential transmission of AA under nonexperimental conditions. Further information on amyloid transmission is provided in another review article. 168
Although AA amyloidosis is a systemic disease, a localized condition known as amyloid arthropathy has been described in chickens (Fig. 3i).132,134 Amyloid arthropathy is secondary to arthritis caused by Enterococcus faecalis or Mycoplasma synoviae infection in the brown layers.50,235 It is unclear whether the deposited AA is derived from circulating or locally produced SAA, although a previous study reported that naive and Lipopolysaccharide-stimulated fibroblast-like synoviocytes isolated from healthy chicken joints produce SAA. 260 Chicken amyloid arthropathy develops into systemic amyloidosis as the disease progresses126,133; thus, it is likely an initial lesion of systemic amyloidosis rather than localized amyloidosis.
Immunoglobulin Light Chain
The immunoglobulin light chain can form amyloid fibrils (AL), resulting in systemic or localized amyloidosis.67,271 AL amyloidosis is associated with clonal proliferation of plasma cells that produce immunoglobulin light chains with the same variable domain sequence. Amyloid deposits are composed of either kappa or lambda immunoglobulin light chains produced by monoclonal plasma cells, which can be detected by immunohistochemistry or mass spectrometry. However, immunohistochemistry may produce negative results if the antibody does not recognize the specific conformation and the neighboring constant domain. 111 Therefore, it is advisable to use multiple antibodies targeting different constant domains of both kappa and lambda light chains.90,111 AL amyloidosis in animals is commonly localized in association with a plasma cell tumor. Systemic AL amyloidosis is rare in animals and has only been reported in a horse with multiple myeloma 118 and a cow with bovine leukocyte adhesion deficiency. 251
Transthyretin
Transthyretin (TTR), also known as prealbumin, is a plasma transport protein that is mainly produced in the liver and plays a role in the transport of thyroxine (T4) and retinol (vitamin A). TTR amyloidosis has been described in vervet monkeys (Chlorocebus pygerythrus).29,176 Interestingly, vervet monkeys harbor an amino acid substitution at amino acid 122 of the TTR protein (Ile122) compared to other primates (Val122). 259 Several types of TTR mutations associated with familial forms of TTR amyloidosis have been reported in humans. The Ile122 allele, the most common TTR mutation in the United States, causes late-onset cardiac amyloidosis. 102 The clinical manifestations and pathology of TTR amyloidosis in vervet monkeys are comparable to those in the human disease. Almost half of vervet monkeys over 14 years of age show signs of cardiac dysfunction, such as arrhythmia, increased cardiothoracic ratio on radiography, increased plasma atrial natriuretic peptide concentration, and reduced ejection fraction. 259 Amyloid deposits are observed in the vascular walls and interstitium of the heart and, to a lesser degree, in other organs, including the intestines, kidneys, lungs, tongue, and adipose tissue (Fig. 4a).29,259
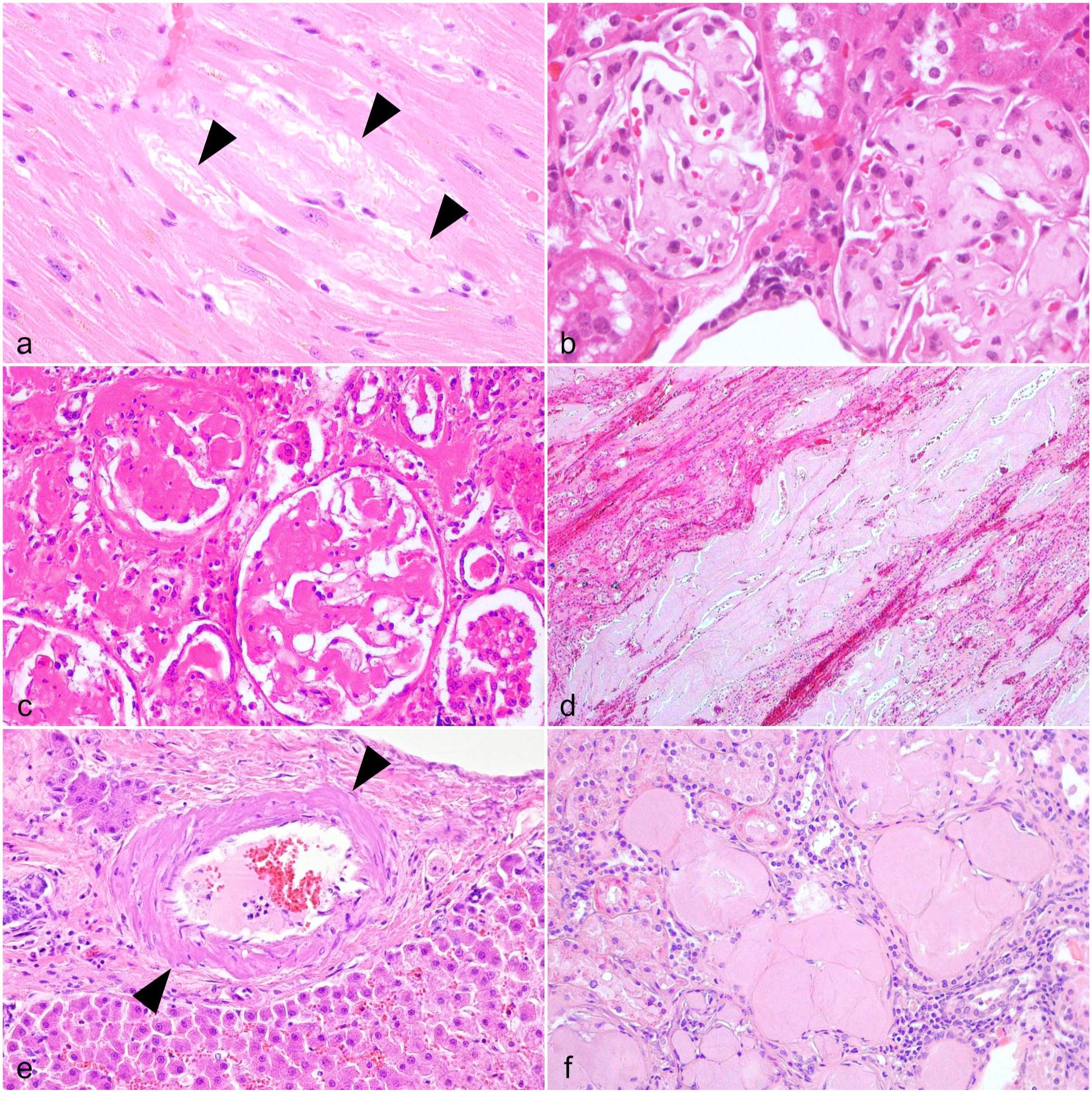
Histologic features of systemic non-AA amyloidosis in animals. Hematoxylin and eosin.
Apolipoprotein A-II
ApoA-II is the second most abundant serum high-density lipoprotein and is mainly produced by hepatocytes. Inbred mouse strains spontaneously develop systemic ApoA-II amyloidosis with aging in the absence of an increase in serum ApoA-II concentration. 86 ApoA-II amyloid deposits affect many extracerebral organs, including the kidney, liver, spleen, heart, lung, gastrointestinal tract, tongue, and skin. Although the prevalence and development of this disease differ among mouse strains, several studies have reported that aged wild-type C57BL/6 mice originating from the Jackson Laboratory (C57BL/6J) exhibit high incidence and progressive organ dysfunction.22,135,222 Glomerular amyloid deposition is associated with podocyte injury and glomerulosclerosis, resulting in albuminuria (Fig. 4b).
There are multiple alleles of murine Apoa2 gene (A1, A2, B, C, D, E, and F), of which A1 and C are associated with ApoA-II amyloidosis predisposition. Compared to wild-type C57BL/6J mice carrying the type-A1 Apoa2 allele, mice carrying the type-C Apoa2 allele, such as senescence-accelerated mouse-prone1 (SAMP1) mice exhibit more severe amyloid deposition throughout the body, whereas Algerian mouse (Mus spretus)-derived laboratory mice (SPRET/Ei mice) carrying the type-F Apoa2 allele are resistant to amyloid deposition.119,221 In addition, genetic analysis of the less amyloidogenic A/J mouse and SAMP1 strains identified 2 susceptible loci in the telomeric regions of chromosomes 4 and 19. 76 An experimental study has shown that administration of tissue-extracted ApoA-II amyloid drastically shortens the period to disease onset. 85 ApoA-II amyloidosis was induced in young mice reared in the same cage as old mice that developed severe ApoA-II amyloidosis. 276 Transmissible amyloid seeds were detected from feces and milk of amyloid-laden mice.129,276 The mechanism by which aging is involved in the progression of ApoA-II amyloidosis has yet to be elucidated.
Apolipoprotein A-IV
ApoA-IV amyloidosis is an uncommon form of systemic amyloidosis, in which ApoA-IV–derived amyloid accumulates in multiple organs.40,147 This condition has not been observed in animals except in one case report of a cotton-top tamarin (Saguinus oedipus). 224
ApoA-IV amyloidosis primarily affects the heart and kidneys in humans, showing distinctive signs of gradually worsening cardiac and renal functions. Notably, amyloid deposition in the kidneys tends to be severe in the medulla and infrequent in the glomeruli.147,167 In the cotton-top tamarin, amyloid was deposited predominantly in the glomeruli (Fig. 4c). In addition to the kidneys, severe amyloid deposition was observed in the liver, spleen, ovaries, adrenal glands, and intestinal lamina propria of the tamarin, which has not been documented in human patients with ApoA-IV amyloidosis.173,224 Because the distribution of amyloid is similar to that of AA amyloidosis in callitrichids, 142 immunohistochemistry is necessary to rule out AA amyloidosis.
It should be noted that ApoA-IV is also an amyloid signature protein in other types of amyloidosis. Therefore, the diagnosis of ApoA-IV amyloidosis should not be based only on the ApoA-IV-positive reaction by immunohistochemistry; quantitative evaluation based on mass spectrometry is essential for diagnosing ApoA-IV amyloidosis. In humans, the diagnostic criteria for ApoA-IV amyloidosis include a high number of ApoA-IV peptides detected by mass spectrometry and the absence or low levels of other amyloidogenic proteins. 36 The diagnosis of animal ApoA-IV amyloidosis should also be based on this criterion.
Apolipoprotein C-III
Apolipoprotein C-III (ApoC-III) is a protein involved in triglyceride-rich lipoprotein metabolism. 202 ApoC-III amyloidosis, in which ApoC-III is the precursor protein, is a rare disease and has only been reported in one report each for humans and animals. In humans, the D25V mutation of ApoC-III is implicated in its etiology. 261 In animals, ApoC-III amyloidosis is reported in white lions (Panthera leo), which, unlike humans, have no mutation in the APOC3 gene. 125 It has been suggested that ApoC-III amyloidosis in lions may be age-related or involve genetic background factors. 125 Histologically, amyloid deposits in the systemic vascular walls and interstitial tissues of the affected white lions (Fig. 4d). Deposition is particularly severe in the kidneys and is confined to the interstitium of the extramedullary zone. The pattern of renal amyloid deposition differs from that of AA amyloidosis, which involves the glomerulus; however, it may be difficult to differentiate them without immunohistochemistry. An anti-human ApoC-III polyclonal antibody (Bioss, Woburn, MA) can be used for immunohistochemistry. 125
EGF-Containing Fibulin-Like Extracellular Matrix Protein 1
EGF-containing fibulin-like extracellular matrix protein 1 (EFEMP1) amyloidosis has been reported in humans 254 and a Tsushima leopard cat (Prionailurus bengalensis euptilurus). 160 Interestingly, in humans, the C-terminal region of EFEMP1 is involved in amyloid formation, whereas in the Tsushima leopard cat, the N-terminal region of EFEMP1 is thought to be involved. Amyloid is mainly deposited in venous walls of humans, whereas it was mainly deposited in the arterial walls of the Tsushima leopard cat, perhaps reflecting different amyloidogenic regions of the protein (Fig. 4e). EFEMP1 amyloidosis in the Tsushima leopard cat can be histologically differentiated from AA amyloidosis by the lack of amyloid deposition in the renal tubular interstitium and severe deposition in the submucosal tissue rather than the lamina propria of the gastrointestinal tract. EFEMP1 is produced locally in the vascular walls, resulting in amyloid deposition in blood vessels and perivascular stroma. Therefore, it can also be considered localized amyloidosis in systemic vessels rather than systemic amyloidosis. 253
Fibrinogen Aα-Chain
Fibrinogen Aα-chain amyloid (AFib) causes a systemic disease in animals and humans.69,279 In animals, AFib amyloidosis has been reported in aged Japanese squirrels (Sciurus lis). 97 In humans and squirrels, the amyloid is exclusively deposited in the glomeruli, resulting in renal failure (Fig. 4f).14,97 Amyloid deposits may also be seen in other organs in severely affected cases.14,97 Although the precursor protein plays a role in coagulation, AFib amyloidosis is not associated with coagulopathy in humans. 63 AFib amyloidosis can be differentiated from AA amyloidosis, which involves the tubulointerstitium, perivascular space, and glomeruli. As only approximately 100 amino acid residues at the C-terminus of AFib are involved in amyloidogenesis, antibodies that recognize this region should be used for immunohistochemistry. For immunohistochemistry, an anti-human AFib monoclonal antibody (clone sc-398806) is recommended. 97
Apolipoprotein A-I
Apolipoprotein A-I (ApoA-I) amyloidosis is hereditary systemic amyloidosis or acquired localized arterial amyloidosis in humans.7,37,164 In animals, localized amyloidosis occurring in the pulmonary vasculature has been reported in dogs.49,108,215 The incidence increased with age, occurring in 22% of dogs older than 10 years. 215 Since there was no effect of ApoA-I mutation, sex, breed, or concomitant diseases, it is speculated that abnormal metabolism of ApoA-I associated with aging is involved in the pathogenesis of the disease. However, no studies on canine ApoA-I amyloidosis have been reported since the 1990s, and the detailed pathogenesis of this disease remains unknown. Since ApoA-I also has the properties of an amyloid signature protein, this should be considered when diagnosing amyloidosis by immunohistochemistry or mass spectrometry. 160
Islet Amyloid Polypeptide
IAPP, also known as amylin, is a hormone linked to type 2 diabetes, which consists of 37 amino acids after posttranslational processing of its original pre-pro-peptide. IAPP-derived amyloid deposition in the pancreas is reported in various animal species, including monkeys, 41 dogs,184,185 and cats. 285 Clinical symptoms of IAPP amyloidosis-affected animals have not been described, and amyloid deposits are often found incidentally at autopsy. 81 Amyloid in islets affects beta cells and the vasculature (Fig. 5a). 27
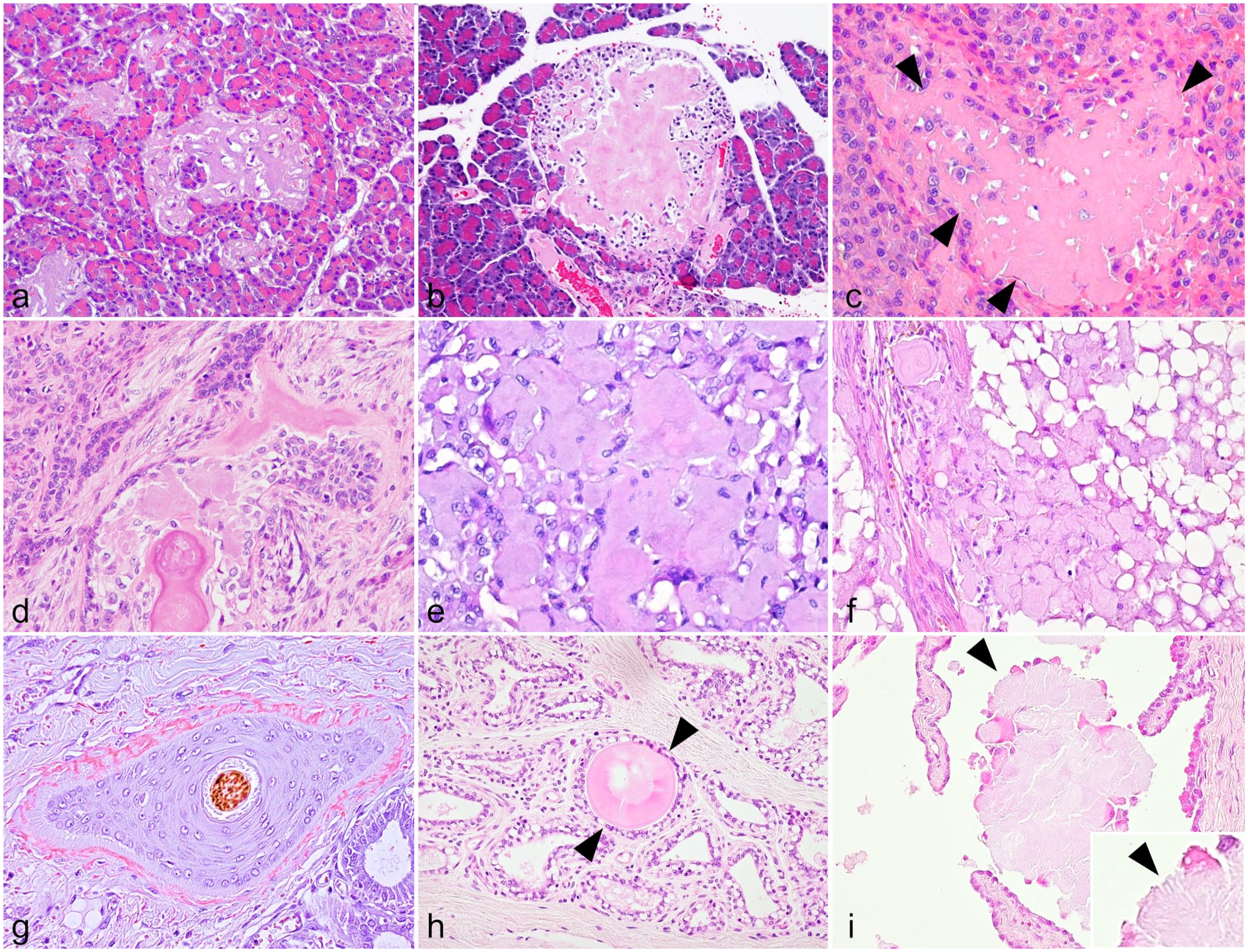
Histologic features of localized amyloid deposition in animals.
More than 70% of feline and human patients with type 2 diabetes have amyloid deposits in the pancreatic islets.93,183,205,272 Diabetic cats exhibit reduced islet size, beta cell loss, and mild inflammation. 285 A recent study investigated the amyloidogenic properties of feline IAPP and its positive relationship with feline diabetes and pancreatitis. 110 Feline IAPP is highly prone to fibril formation, similar to human diabetes-associated IAPP. Elevated plasma IAPP levels were detected in feline diabetes patients. 110 One functional role of IAPP is to provide satiety. IAPP is stored in the secretory granules of beta cells. In type 2 diabetes, secretion of IAPP and insulin demand is elevated, increasing local concentration of IAPP, which may result in amyloid depositions. 68
There are variations in the amino acid sequence of IAPP among animal species. The presence of 3 prolines in the amino acids 24–29, such as those in IAPP of the rat and mouse, prevents its aggregation, and thus rats and mice do not develop pancreatic amyloidosis. Other species that are known to be resistant to pancreatic amyloidosis are guinea pig, degu (Octodon degus), cow, golden lion tamarin (Leontopithecus rosalia), alpaca (Vicugna pacos), and common bottlenose dolphin (Tursiops truncatus).54,158 Cases of naturally occurring pancreatic amyloidosis without reported prediabetic and/or diabetic status have been reported in nonhuman primates, 24 domestic cats, 24 cougars (Felis concolor), 109 ocelots (Leopardus pardalis), 115 and raccoons (Procyon lotor).24,280 Deposition of IAPP amyloid has been reported in cases of insulinomas in dogs.184,185 Based on an in vitro study, sheep and goats seem resistant to developing type 2 diabetes despite the presence of amyloid deposits in their pancreas. 157
Insulin
Insulin amyloidosis is a phenomenon in which amyloid fibrils derived from insulin are deposited in pancreatic islets (Fig. 5b). 237 In humans, iatrogenic pathology is common and is caused by repeated insulin administration to the same site in the subcutaneous tissue of diabetic patients. This is the preeminent predisposing factor for local amyloid deposition.6,237 In animals, amyloid deposition is commonly observed in the pancreatic islets of aging degus (Fig. 5b). 233 Unusually in this species, the amyloid fibrils appear to be composed of insulin itself, rather than IAPP as observed in the other species studied to date. 271 The onset of the disease in degus seems to be age-related. 84 There are differences in the amino acid sequence of insulin among degus and other animals, including humans, which may account for the structural differences in the protein. 84
Amyloid β
Amyloid β (Aβ) is produced by the cleavage of APP, an integral membrane protein expressed in neurons and other cells. Several Aβ subtypes are produced according to the cleavage site. The major subtypes are Aβ40, Aβ42, and Aβ43. 243 Aβ deposits can be found in the neuropil of the cerebral gray matter and vascular walls of the meninges and cerebrum in aged individuals (Fig. 6a, b). Aβ deposition has been reported in many animal species with high amino acid sequence identity to human Aβ, such as primates, dogs, bears, sea lions, camels, and birds. 282 In such species, Aβ deposits can be detected immunohistochemically using anti-Aβ antibodies raised against human Aβ with formic acid antigen retrieval.30,243
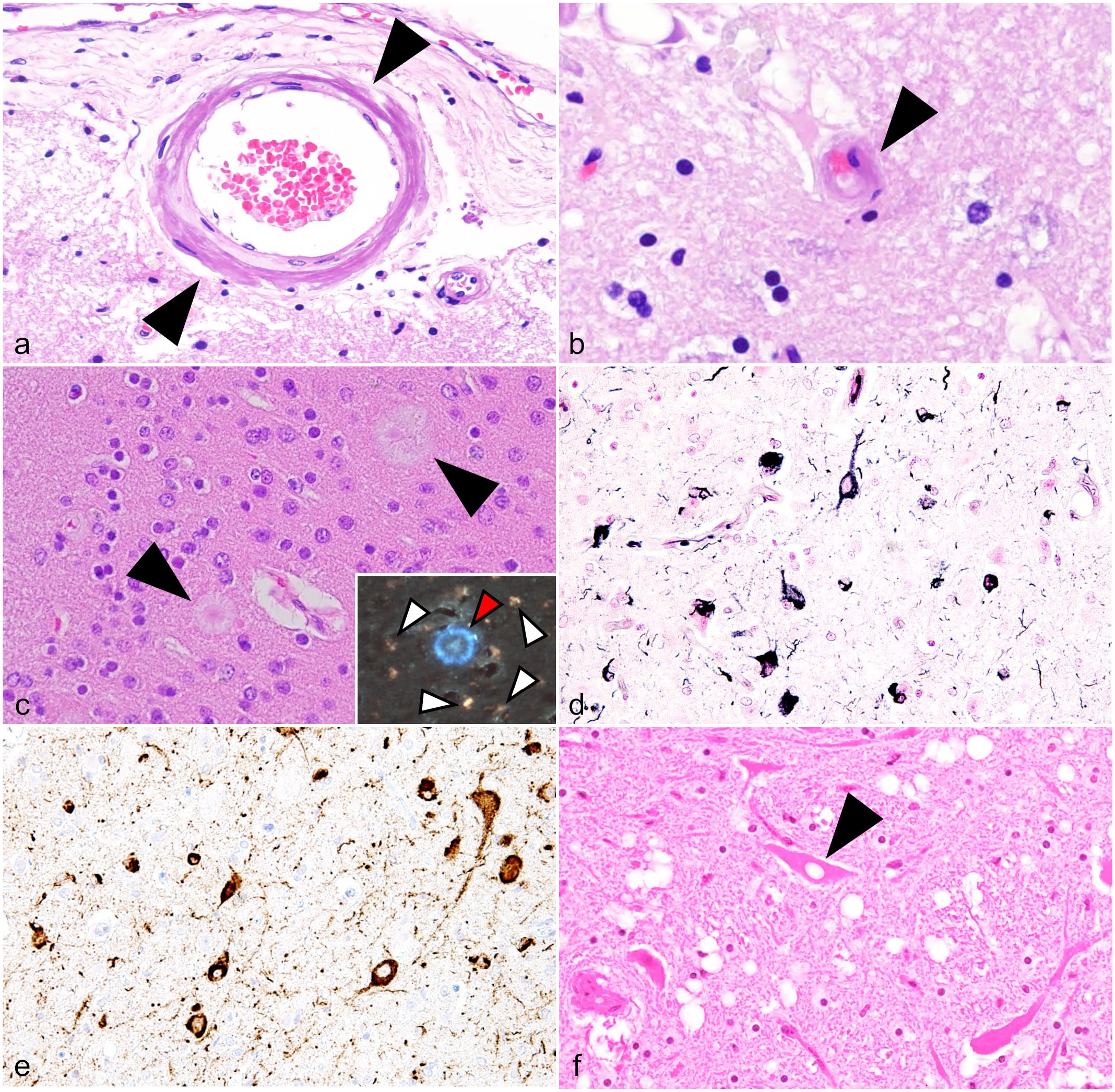
Central nervous system amyloid deposits and related histopathologies in animals.
Aβ deposits in the neuropil are called “senile plaques” and are further classified into mature, diffuse, and other types. Mature plaque, which is also called “classic cored plaque,” has a central core that is Congo red-positive (Fig. 6c), while diffuse plaques are Congo red-negative, suggesting that these two types have different fibrillation properties and may be sequential lesions of a maturation process. 216 Periodic acid-methenamine silver staining is useful for detecting both types of plaques. 281 Senile plaques are associated with Alzheimer’s disease in humans, and a significant number of mature plaques surrounded by dystrophic neurites are formed in the patient’s brain. Mature plaques are rarely observed in animal brains, and the clinical significance of senile plaques in animals is yet to be elucidated. 192 Most Aβ plaques in animals cannot be detected in hematoxylin and eosin–stained sections, and immunohistochemistry or silver staining is necessary. However, Aβ lesions in squirrel monkeys (Saimiri boliviensis) 182 and APP/PS1 transgenic mice 61 exhibit blue-white autofluorescence under blue-violet excitation and can be detected without labeling using fluorescence microscopy (Fig. 6c).
Vascular wall Aβ deposits are involved in cerebral amyloid angiopathy. Aβ deposits are detected in arteries, arterioles, veins, venules, and capillaries using immunohistochemistry. In aged dogs, capillary deposits are distributed multifocally in the cerebral cortex. 31 Cerebral amyloid angiopathy has been associated with microhemorrhage and white matter changes in humans and dogs.33,34,57,257
The sequence of Aβ in felids is 1 amino acid different from human Aβ at the N-terminus (felid-type Aβ).31,33,282 Aged felids develop unique diffuse granular deposits in the cerebral cortex that can be detected by immunohistochemistry using anti-human Aβ antibodies raised against the C-terminus. 53 Also, intraneuronal Aβ accumulation has been reported in domestic cats.31,232 Amino acid sequence of Aβ in mice and rats (rodent-type Aβ) differs by 3 amino acids from the human Aβ, and these animals do not develop spontaneous amyloid deposits in their brains.
Tau
Tau is a microtubule-associated protein that functions in the assembly and stabilization of microtubules in the neuronal axons. As a simplification of the 6 known tau isoforms, 2 major splicing isoforms are distinguished by the number of repeats in the microtubule-binding domain: 3-repeat (3R) tau and 4-repeat (4R) tau. 252 Intracellular aggregation of misfolded tau protein is responsible for a group of neurological diseases in humans called “tauopathies,” which includes Alzheimer’s disease, progressive supranuclear palsy, corticobasal degeneration, chronic traumatic encephalopathy, and others. Affected cells (neurons, astrocytes, and oligodendrocytes), aggregated tau isoforms, and distributions differ between these diseases. 252 As described before, misfolded tau can be found extracellularly upon cell death or in the synaptic cleft, and its cytotoxity is due to aggregation; therefore, the misfolded tau has recently been classified as an amyloid.24,230
Alzheimer’s disease is the most prevalent tauopathy in humans and is histopathologically characterized by neurofibrillary tangles (NFTs), which are fibrillar aggregates of hyperphosphorylated tau protein in the cerebral neurons. NFTs are composed of both 3R and 4R tau. NFT is detected by Gallyas-Braak silver staining (Fig. 6d) and is immunohistochemically positive using anti-hyperphosphorylated tau antibodies such as clones AT8 (Ser202, Thr205) and AT100 (Thr212, Ser214; Fig. 6e). Aged felids, including domestic cats, cheetahs, and leopard cats, are known to develop significant numbers of NFTs comparable to those in Alzheimer’s disease.31,32,53,75,83,226,232 NFTs have been reported in other animal species, including monkeys and pinnipeds.45,198,246 Hyperphosphorylated tau-positive neurons have been observed in the brains of headbutting bovids, which may be associated with chronic traumatic encephalopathy, a neurological condition described in American football players and people who have experienced repeated head trauma. 3 Misfolded hyperphosphorylated tau and its spreading occur predominantly in stages 3 and 4 of human chronic traumatic encephalopathy (in Dr. McKee’s staging scheme).52,152 NFTs are rarely found in the brains of aged dogs (personal communication by an author, JKC). In addition to NFTs, hyperphosphorylated tau aggregates in astrocytes, which are characteristic of progressive supranuclear palsy and corticobasilar degeneration in humans, have been reported in aged cynomolgus monkeys (Macaca fascicularis).117,258 Hyperphosphorylated tau aggregates are also found in oligodendrocytes of aged felids, pinnipeds, and monkeys.31,246,258 The clinical significance of tauopathy in animals is yet to be elucidated.
Wild-type mice do not develop NFTs, which is likely attributable to the expression patterns of the tau isoforms. Both 3R and 4R isoforms are expressed in the adult human brain, whereas only the 4R isoform is expressed in the mouse brain. 248 The ratio of 3R to 4R isoforms expressed in animal brains varies among species.31,104,246
α-Synuclein
α-Synuclein is mainly expressed in the axon terminals of presynaptic neurons and plays a role in neurotransmitter release. 238 In humans, intracellular aggregation of misfolded α-synuclein protein is involved in Parkinson’s disease, dementia with Lewy bodies, and multiple-system atrophy, which are collectively called “synucleinopathies.” Neuronal inclusions composed of misfolded α-synuclein, known as Lewy bodies, are the characteristic lesions of Parkinson’s disease and dementia with Lewy bodies. Lewy bodies have not been reported in animals.
Aggregation of α-synuclein can be induced in wild-type mice and monkeys by intracerebral injections of α-synuclein extracted from the brains of human patients with synucleinopathy, suggesting the prion-like nature of α-synuclein.149,206 In addition, α-synuclein aggregates have been reported in transgenic mouse strains that overexpress the human hyperphosphorylated-tau protein.244,245 These experimental studies suggest that native mouse and monkey α-synucleins have the potential to aggregate by seeding with exogenous amyloid proteins. Seeding is thought to be involved in intracerebral propagation of misfolded α-synuclein.139,179,191
In veterinary medicine, misfolded α-synuclein occurs in aged horses affected with pituitary pars intermedia dysfunction.55,151 Higher levels of α-synuclein have been detected by McFarlane et al 151 in pituitary pars intermedia dysfunction horses. Seeding experiments and electron microscopic studies demonstrated the presence of misfolded α-synuclein in horses with pituitary pars intermedia dysfunction compared to aged and young horses. 55 Several laboratories have studied the aggregation kinetics (or propensity to misfold) of α-synuclein peptides designed from amino acid sequences of animal species.56,220 Further studies are needed to determine whether spontaneous aggregation of α-synuclein occurs in additional animal species.
Prion Protein
Prion diseases or transmissible spongiform encephalopathies are neurodegenerative disorders in which the conformation of cellular prion proteins (PrPC) becomes abnormal, and their aggregates accumulate in the brain, resulting in functional impairment. 94 Prion diseases have been considered protein misfolding disorders, but like other amyloidoses, 174 they have in vitro seeding activity and the ability to form fibrils and can be considered a type of amyloidosis in a broad sense. 284 Prion diseases have been reported in primates, sheep, goats, minks, felids, camels, deer, cows, and other exotic ruminants.190,227 Vacuolar changes and aggregation of misfolded prion protein (PrPSc) in the brain are histopathological characteristics regardless of species (Fig. 6f).
Conformational changes in PrPC can be induced by the transmission of PrPSc, which is mostly the case in animals; by an inherited mutated prion gene (PRNP); or spontaneously in atypical cases.4,73 In the brain, PrPSc deposits can be detected intracellularly (neurons and glial cells) or extracellularly using immunohistochemistry with formic acid and heat antigen retrieval. 263 From a diagnostic perspective, amyloid plaques may be recognized on hematoxylin and eosin-stained sections; however, they are more easily detected by silver staining, Congo red staining, and most dramatically, by immunohistochemistry.74,273
Scrapie is the oldest known prion disease and occurs in sheep and goats. Clinical manifestations include tremor and behavioral changes, but intense pruritus is the most characteristic clinical sign.73,94 The incubation period for scrapie is 2–5 years. 94 PrPSc has been detected in the nervous system, pancreas, heart, skin, bladder, mammary gland, and saliva.64,73 After oral uptake, pathogenic PrPSc crosses the intestinal mucosal barrier, replicates in gut-associated lymphoid tissues, and then spreads to the brain via neuronal or hematogenous pathways.247,263 Atypical scrapie, which differs in pathological features compared to typical scrapie, has been reported.13,120,150,158 Although the distribution of PrPSc deposits in the brain varies among animal species and disease types, the medulla oblongata is the most consistent diagnostic region for transmissible spongiform encephalopathies, except in atypical prion diseases, which mainly involves cerebellum.13,65 Atypical scrapie spontaneously occurs most commonly at 5 years of age and older and is thought to occur sporadically with age rather than by transmission. 2 The main clinical manifestations of atypical scrapie are ataxia and dyscoordination, but pruritus is rare. 94
Chronic wasting disease (CWD) is a prion disease found in captive and free-ranging Cervidae family members, mainly in North America, and eradication is a major challenge. 94 Although the detailed pathogenesis remains unknown, horizontal transmission is suspected; CWD-like spongiform encephalopathy can be induced by intracerebral inoculation of sheep PrPSc in elk. 79 Since the pathogenic prion protein (PrPCWD) is detected in gut-associated lymphoid tissues,228,234 prions may be shed into the environment via feces and saliva. In addition, CWD prions can remain pathogenic in soil for long periods of time, which may contribute to sporadic disease outbreaks.106,107 PrPCWD is found in various organs of affected animals, like scrapie prion. 94
Bovine spongiform encephalopathy is a prion disease that occurs in cattle and is associated with clinical signs such as tremors and abnormal gait of the hind limbs. 94 The incubation period of bovine spongiform encephalopathy is 2–8 years, and most cases occur at the age of 4–5 years. 181 PrPSc accumulation is usually seen in the brain, but bovine spongiform encephalopathy prions may also be detected in the spinal cord, retina, adrenal glands, ileum, tonsils, bone marrow, peripheral nerves, ganglia, and muscle in late stages of the disease.94,187
Distinct strains of prions have been identified and associated with different incubation periods, clinical signs, tissue tropisms, and host ranges.11,145 In addition, host PRNP genotypes have been associated with a predisposition to resistance or susceptibility to prion disease.2,94,190 For example, PRNP gene mutations are involved in the pathogenesis of atypical scrapie.38,73 Variations in host genotypes and prion strains result in complex and diverse disease phenotypes, which is beyond the scope of this review. 65
Tumor-Associated Localized Amyloidosis
Amyloid deposits may occur in tumors that produce amyloid precursor proteins. In animals, tumor-associated localized amyloidosis has been reported in extramedullary plasmacytomas, amyloid-producing ameloblastomas, C-cell carcinomas, insulinomas (covered above in the IAPP section), and mammary gland tumors.
Plasmacytoma
In extramedullary plasmacytomas, monoclonal immunoglobulin (M protein) produced by neoplastic cells may form amyloid. 5 As described previously, in systemic AL amyloidosis, amyloid is derived from free light chains in the serum, resulting in vascular and perivascular amyloid deposits. 5 Conversely, locally produced light chains form nodular amyloid deposits in plasmacytoma-associated localized AL amyloidosis (Fig. 5c). 270 Amyloid deposits can also be seen in the vessels surrounding the amyloid nodules, but they are distinguishable from systemic amyloidosis because the vascular deposits disappear away from the central lesion. 111 Granulomatous inflammation characterized by multinucleated giant cells commonly accompanies localized AL amyloidosis.111,116,204
Since AL amyloid is largely constituted from the variable domain of the light chain, diagnosis of AL amyloidosis by immunohistochemistry is challenging as described in the AL amyloidosis section above. 111 Examination of the protein components by mass spectrometry supports the diagnosis. 111 Practically, morphological diagnosis of plasmacytoma, supported by immunohistochemistry for plasma cell markers such as MUM1, 80 strongly suggests that the locally deposited amyloids are AL. Immunoglobulin light chains are classified into kappa and lambda light chains, but all localized AL amyloidoses in animals identified to date are derived from the lambda light chain.80,111,116,138,204
It has been suggested that AL amyloid is toxic to tumor cells. 270 In some cases, the tumor cells die out, and only the amyloid nodule is left.111,116,138,204 In humans, although it is not known whether amyloid-induced tumor cell death is involved, plasma cell tumors with localized AL amyloidosis has a better prognosis than plasma cell tumors without amyloid. 146 In the author’s experience, cutaneous plasma cell tumors with amyloid in dogs have a good prognosis, although multiple lesions may develop. Plasma cell tumors with amyloid in visceral organs, mostly reported in cats, 219 have a poor prognosis because of organ dysfunction.240,249 Investigating the prognostic impact of amyloid deposition on tumor outcomes in animals may provide clinically useful information.
Amyloid-Producing Ameloblastoma
Amyloid-producing ameloblastoma has been reported in cats,19,43,62,186 dogs,62,89,186 a goat, 140 a prairie dog (Cynomys ludovicianus), 128 and a Bengal tiger (Panthera tigris tigris). 114 This tumor in animals used to be called amyloid-producing odontogenic tumor but was renamed “amyloid-producing ameloblastoma.” 44 Histologically, these tumors are characterized by irregular nests, islands, and strands of proliferating odontogenic epithelium supported by varying amounts of fibrous stroma with moderate to severe amyloid deposits (Fig. 5d). 148
Amyloid-producing ameloblastoma was previously considered equivalent to calcifying epithelial odontogenic tumor in humans; however, the current consensus is that amyloid-producing ameloblastoma is not analogous to calcifying epithelial odontogenic tumor.18,101 In calcifying epithelial odontogenic tumor, odontogenic ameloblast-associated protein has been identified as the amyloid precursor protein. 24 However, ameloblastin is a major component of amyloid deposits in amyloid-producing ameloblastoma by mass spectrometry–based proteomic analysis, and immunohistochemistry has shown evidence of the presence of the N-terminal region of ameloblastin in amyloid deposits in both cats and dogs.87,89,148 Ameloblastin is a protein found in the enamel, the outer layer of the tooth, and is produced by differentiated dental epithelial cells known as ameloblasts.105,163 It is necessary to select antibodies that react with the N-terminus of ameloblastin for its detection in tissue sections by immunohistochemistry.
C-Cell Carcinoma
Amyloid deposition occurs in 80% of human thyroid C-cell carcinomas, and calcitonin has been identified as the precursor protein. 46 On the other hand, amyloid deposition is uncommon in animal C-cell carcinomas and has been reported sporadically in dogs,196,197 foxes, 88 sheep, 207 bulls, 17 and a cat (Fig. 5e). 170 CRSP1 was found to be the amyloid precursor protein in feline C-cell carcinomas. 170 Considering the high identity of CRSP1 among cats and canines, it is likely that CRSP1 also forms amyloids in canine and fox C-cell carcinomas. 170 In contrast, bovine calcitonin has amyloidogenic properties in vitro, 266 and it is possible that the amyloid precursor protein of amyloid-producing C-cell carcinomas in ruminants is calcitonin, similar to that in humans.
Mammary Gland Tumors
Canine mammary tumors may be associated with localized amyloidosis characterized by diffuse to nodular amyloid deposits in the stroma (Fig. 5f).250,255,264 Amyloid deposits should be differentiated from corpora amylacea, which form in the mammary gland ducts. Mass spectrometry–based proteomic analysis has identified α-S1-casein as the amyloid precursor protein. 169 Amyloid-producing canine mammary tumors overexpress α-S1-casein, and the N-terminal region is truncated in α-S1-casein within amyloid deposits, suggesting that α-S1-casein acquires amyloidogenicity through supersaturation-associated self-aggregation and proteolysis.71,169 Given the propensity of various milk proteins to form amyloids in vitro,48,78,194,199,256 mammary tumor–associated amyloidosis may also occur in other species.
Amyloid Deposition of Unknown Pathological Significance
This section summarizes amyloid deposits of unknown pathological significance in animals.
Keratinic Amyloid Deposition in the Skin
Amyloid deposition is occasionally observed in hair follicle tumor’s stroma and around normal hair follicles in dogs (Fig. 5g). 124 Although this condition appears to correspond to keratinic primary localized cutaneous amyloidosis in humans, 156 its pathological significance remains unknown because of the negligible amount of amyloid deposition found in dogs. Keratin 5 has been identified as the precursor protein of amyloid formation. 124 However, the mechanism underlying how keratin 5 expressed in keratinocytes deposits under the basement membrane to generate amyloid fibrils is unknown. The amyloid deposits in these cases can be immunolabeled using the anti-keratin 5 monoclonal antibody clone XM26. 124
Amyloid Deposition in the Corpora Amylacea in Mammary Glands
Corpora amylacea are periodic acid-Schiff-positive spherical structures that may be observed in various organs.28,113,213,275 In 1854, Rudolph Virchow discovered iodine-reactive spherical structures in the brain and named them corpora amylacea, from which the term “amyloid (starch-like)” originated. 130 It should be noted, however, that the Latin term corpora amylacea, which means amyloid body, does not necessarily contain amyloid. In cattle, 180 rats, 12 and dogs, 250 corpora amylacea formed in the mammary gland often contain amyloid; α-S2-casein has been identified as an amyloid precursor protein in cattle (Fig. 5h). 180
In the mammary glands of aged rats, needle-like amyloid deposits are observed on the surface of the corpora amylacea (Fig. 5i). 172 Mass spectrometry–based proteome analysis has identified lipopolysaccharide-binding protein as a precursor protein of needle-like amyloid, and synthetic peptides derived from lipopolysaccharide-binding protein actually form amyloid in vitro. 172 Inflammatory cell clusters are observed around needle amyloid deposits.
Although corpora amylacea were discovered long ago, their pathological or functional significance is still under debate. In recent years, the concept of “wasteosomes” has emerged. 209 The mammary gland has a potentially high risk of protein misfolding owing to its high protein production and concentration. Indeed, many milk proteins, such as α-S1-casein and κ-casein, have amyloid-forming properties that are often cytotoxic.42,170 Corpora amylacea (acting as wasteosomes) may have a protective function by trapping pathologically misfolded proteins within their glycan structures and safely metabolizing or releasing them. In fact, corpora amylacea in the human brain contain ubiquitinated waste products, which are thought to contribute to brain clearance systems by expelling them into the cerebrospinal fluid.208,210
Amyloidosis in Vertebrates Other Than Mammalian and Avian Species
Amyloidosis in vertebrates other than mammalian and avian species is extremely rare, and many reports have failed to prove the presence of amyloid in histologically suspicious lesions. However, at least two papers have demonstrated amyloid deposition.23,25 In the African tiger snake (Telescopus semiannulatus), massive amyloid (confirmed by congophilia, fluorescence of a luminescent-conjugated oligothiophene [h-FTAA] probe and transmission electron microscopy) was deposited in the interstitium and vessel walls of the spleen and testis. 23 Amyloid was negative on anti-SAA immunolabeling, and the amyloid precursor protein is unknown. 23 In the stingray (Bathytoshia centroura), amyloid-like structures were deposited in the renal mesangium. 25 Although deposits were negative for Congo red and thioflavin T, electron microscopy revealed amyloid-like fibrils. 25 Several other cases of suspected amyloidosis in vertebrates other than mammalian and avian species have been reported, but none have formally confirmed amyloid by additional methods, and these are not discussed further. Although there have been no known reports of amyloidosis in invertebrates, it is worth noting that bacteria, fungi, and insects (and even humans) use amyloid to achieve diverse biological functions (the so-called functional amyloid).21,47
Conclusion and Perspectives
As the pathogenesis of amyloidosis differs depending on the amyloid precursor protein, amyloid typing is important to understand its pathogenesis. To date, 21 amyloid precursor proteins have been identified in animals. Some of these have been reported only in specific animal species and are likely to be associated with species-specific protein amino acid sequences and metabolism.84,97,161,224,259 In contrast, aging is a predisposing factor for amyloidosis across species.45,84,97,135,215,246,253 With the increasing longevity of captive animals, it is highly possible that novel amyloidoses will be found in the future. However, the significance of age-related amyloid deposition, such as whether it is a pathological change that causes organ damage and dysfunction or a physiological change associated with age-dependent accumulation of misfolded proteins, is often unclear in animals. 253 Further comparative pathological studies of the amino acid sequence and nature of amyloids may enable an evolutionary understanding of physiological protein aggregation and protein misfolding diseases.
The authors hope that the diagnostic methods and classifications described here will help veterinary pathologists in diagnostic practice. Moreover, novel amyloids that are not listed here are likely to be discovered by veterinary pathologists. The classification of animal amyloidosis should be updated periodically by gathering more information on its pathogenesis and clinical features.
Footnotes
Acknowledgements
The authors thank Prof. Shinichiro Nakamura for providing HE-stained tissue images of the heart of a velvet monkey. The authors would like to acknowledge Naseem H. Alfadhl for putting together the initial summaries about the animal and experimental studies for the human IAPP section.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.