
Abstract
Emerging and re-emerging human coronaviruses (hCoVs) cause severe respiratory illness in humans, but the basis for lethal pneumonia in these diseases is not well understood. Alveolar macrophages (AMs) are key orchestrators of host antiviral defense and tissue tolerance during a variety of respiratory infections, and AM dysfunction is associated with severe COVID-19. In this study, using a mouse model of Middle East respiratory syndrome coronavirus (MERS-CoV) infection, we examined the role of AMs in MERS pathogenesis. Our results show that depletion of AMs using clodronate (CL) liposomes significantly increased morbidity and mortality in human dipeptidyl peptidase 4 knock-in (hDPP4-KI) mice. Detailed examination of control and AM-depleted lungs at different days postinfection revealed increased neutrophil activity but a significantly reduced MERS-CoV-specific CD4 T-cell response in AM-deficient lungs during later stages of infection. Furthermore, enhanced MERS severity in AM-depleted mice correlated with lung inflammation and lesions. Collectively, these data demonstrate that AMs are critical for the development of an optimal virus-specific T-cell response and controlling excessive inflammation during MERS-CoV infection.
Keywords
Human coronaviruses (hCoVs) cause pneumonia of varying degrees of severity. Seasonal low-pathogenic hCoVs (NL63, 229E, OC43, and HKU1) infect the upper respiratory tract causing mild disease, with severe pneumonia occasionally observed in the elderly and immunocompromised individuals.45,55,70,73 Highly pathogenic hCoVs such as severe acute respiratory syndrome coronavirus (SARS-CoV), Middle East respiratory syndrome coronavirus (MERS-CoV), and recently emerged SARS-CoV-2 infect both upper and lower airways, causing moderate to severe pneumonia with fatal pneumonia observed primarily in aged or immunocompromised individuals or those with co-morbidities.68,79–81,88 MERS-CoV emerged in the Middle East in 2012 and has since spread to 27 countries infecting more than 2500 people with 858 known deaths.47,49,68,80 Although the total number of infections and deaths caused by MERS-CoV is less than that caused by SARS-CoV-2, continuous circulation of MERS-CoV in camels and isolation of several MERS-like CoVs in camelids and other intermediate hosts poses a risk for species crossover and a threat to human health.1,2,3,12,19,67
Middle East respiratory syndrome symptoms, typical of other highly pathogenic hCoVs, include high fever, cough, and shortness of breath.6,7,8,60 Atypical symptoms such as diarrhea and asymptomatic infections are also common during MERS and other hCoV infections.8,60 Middle East respiratory syndrome coronavirus infects type-I and -II pneumocytes, nonciliated bronchial epithelial cells, endothelial cells, and some hematopoietic cells, using dipeptidyl peptidase 4 as a receptor to enter cells.13,37 Fatal pneumonia caused by hCoVs, including MERS-CoV, is characterized by rapid virus replication to high titers, impaired and delayed antiviral interferon (IFN)-stimulated gene (ISG) responses, excessive myeloid cell inflammatory activity, and lymphopenia.57,59,74 Such an immune response, often referred to as “dysregulated immunity,” causes impaired virus clearance, an exaggerated inflammatory response, acute lung injury caused by direct virus cytopathic effects and inflammatory mediators, acute respiratory distress syndrome and death.14,15,57,59,74 Another key host protective mechanism, besides antiviral immunity, is “host tolerance” to virus-induced inflammation and tissue injury.4,9,10,54,62 Several preclinical and clinical studies identified a variable disease outcome that ranges from mild or moderate respiratory illness to fatal pneumonia despite comparable hCoV load in the airways, lungs, and blood.36,43,44,53 These results highlight the role of host tolerance in SARS, MERS, and COVID-19 outcomes.
Alveolar macrophages (AMs) are key immune cells and are part of the initial antiviral response against a variety of pneumotropic viruses.29,51,65,77 Alveolar macrophages sense invading viruses via cell surface/endosomal Toll-like receptors and/or intracellular sensors and produce elevated levels of antiviral IFNs/ISGs to protect the host from viruses such as influenza A virus, respiratory syncytial virus, and Sendai virus.30,51 However, the role of AMs during hCoV pathogenesis appears to be virus-specific. In contrast to their IFN/ISG-mediated antiviral role during several viral lung infections,30,38,51,65,77,78 AMs fail to produce IFNs and ISGs during hCoV infections such as SARS-CoV-2 and hCoV-229E.24,29 Alveolar macrophages also suppress migration of lung dendritic cells to draining lymph nodes, leading to suboptimal SARS-CoV-specific T-cell immunity. 86 Apart from orchestrating an antiviral response, AMs regulate the influx of myeloid cells into the lungs, moderate lung inflammation, and promote resolution of inflammation and tissue repair, thus facilitating host tolerance to pathogen-induced tissue damage.5,22,26,27,61 However, the role of AMs in the antiviral response and/or host tolerance to MERS-CoV infection is not known.
To study the role of AMs in MERS pathogenesis, we used hDPP4-KI mice infected with MERS-CoV-MA. Based on the literature and our previous studies, we hypothesized that hCoV-specific differential regulation of innate and adaptive immunity by AMs is one of the key determinants of MERS outcomes. Therefore, the objective of this study was to elucidate the role of AMs in pathogenesis and the host response to MERS-CoV infection. We show that CL-liposome-mediated AM depletion caused increased morbidity and mortality in MERS-CoV-infected mice. Alveolar macrophage depletion also facilitated differential myeloid cell activity and impaired MERS-CoV-specific T-cell responses. Furthermore, we observed increased lung pathology in hDPP4 mice lacking AMs. Collectively, our results suggest that AMs have a protective role in MERS and highlight a central role played by AMs in MERS-CoV pathogenesis.
Materials and Methods
Mice, Virus, and Cells
Wild-type (WT) human isolate of MERS-CoV fails to infect mice because the dipeptidyl peptidase 4 (DPP4, the MERS-CoV receptor) of murine origin does not support virus entry. As a result, we developed a human DPP4 knock-in mouse (hDPP4, Dpp4tm1.1 [DPP4]Pbmj) 48 that was susceptible to WT MERS-CoV infection but did not develop severe disease. To further develop a virus isolate that caused severe pneumonia, we serially passaged the WT isolate of MERS-CoV in hDPP-KI lungs. After 30 passages, we obtained a mouse-adapted (MA) strain of MERS-CoV that caused lethal disease in mice. Details of the MA strain of MERS-CoV (MERS-CoV-MA) are described in our previous publication. 52 Mouse-adapted strain of MERS-CoV-MA was propagated in Huh7 cells in T75 flasks (Corning) using complete Dulbecco’s Modified Eagle’s Medium (DMEM) (supplemented with 10% fetal bovine serum [FBS], 1% penicillin-streptomycin, 1% L-glutamine, 1% sodium bicarbonate, 1% sodium pyruvate, and 1% non-essential amino acids). Huh7 cells were infected at an MOI of 0.1 and at 24-hour postinfection, at an approximately 70% cytopathic effect, cells were freeze-thawed, and small aliquots of clarified cell lysates titered on Vero-81 cells (ATCC: CCL-81) using standard plaque assay protocol as briefly described in “virus titers” section.
Mice Infection and Clodronate (CL)-Liposome Treatment
Male hDPP4-KI mice, 8- to 14-week old, were treated with a single dose of CL-liposome (Liposoma BV, catalog # SKU-C005; 75 µL/mouse via intranasal route) 1 day before infection and an equal amount of 1× phosphate-buffered saline (PBS) was given to control mice via the intranasal route. We recently showed that both male and female mice are equally susceptible to MERS-CoV infection, 52 and therefore, we used male mice as representative of both sexes. Mice of comparable body weight were randomly allocated to each group and 3 to 5 mice per group per experimental replicate were used in these studies. Control and AM-depleted mice were challenged with 150 to 300 PFU of MERS-CoV-MA via the intranasal route in 50 µL DMEM under xylazine (12.5 mg/kg)/ketamine (87.5 mg/kg) intraperitoneal anesthesia. The virus inoculum dose of 150 to 300 PFU causes similar morbidity and mortality in hDPP4-KI mice. We used an identical dose of virus inoculum to infect all mice in control and CL-liposome-treated groups of a particular experimental replicate. For survival studies (Fig. 2), mice were weighed and examined daily for 12 days to assess clinical signs such as respiratory rate, posture/movement, fur status/ruffled fur, and the ability to eat and drink. A weight loss of 30% of the initial body weight or signs of severe clinical illness were considered as an endpoint and mortality, and mice were humanely euthanized by isoflurane overdose. All procedures involving animals were approved by the University of Iowa Office of the Animal Care and Use Committee and Oklahoma State University Animal Care and Use Committee under American Veterinary Medical Association guidelines. To evaluate the myeloid cell and T-cell responses in the lungs of control and CL-liposome-treated mice, groups of mice were euthanized at 2 dpi (myeloid cell response) and 7 dpi (myeloid cell and T-cell response) (Figs. 3 and 4). Cohorts of mice were also euthanized at 2, 4, and 7 dpi to examine the changes in lung histopathology in all mice of control and CL-liposome-treated groups (Figs. 5 and 6).
Bronchoalveolar Lavage Fluid Collection
Mice were euthanized using isoflurane overdose followed by cervical dislocation and were fixed on a styrofoam board by pinning limbs. After properly securing mice, a 1-cm incision was made on the neck near the trachea. The exposed trachea was slightly lifted by placing a 200-µL pipet tip under the trachea. A 0.25-cm incision was made along the length of trachea and 1 mL of PBS was injected using 1 mL syringe/22G needle attached to a plastic tubing. Phosphate-buffered saline was slowly flushed back and forth 3 times, and after the final flush, the fluid was collected from the lungs. Of note, lung used for BAL collection were not used for histopathology studies.
Virus Plaque Assay
Middle East respiratory syndrome CoV titers were determined as described previously. 21 Briefly, lungs collected in PBS were homogenized using Fisher Scientific disposable tissue grinders (cat #02-542-07). Lung homogenates were freeze-thawed and 200 µL of 10-fold serially diluted sample was added to each well of a 12-well plate in duplicate and incubated in 37C/5% CO2 cell culture incubator with brief rocking every 10 min. After 1-h incubation, virus inoculum was removed, wells/cells were washed with PBS to remove unbound virus and the cell monolayer was overlayed with 1 mL of 1:1 mixture of 1.2% agarose and 2X-DMEM. Plates were incubated in 37°C/5% CO2 cell culture incubator for 3 days. After 3 days of incubation, cells were fixed with 10% paraformaldehyde for 30 min, followed by agarose removal, and crystal violet (0.1%) staining.
FACS Cell Phenotype Study and Antibodies
For phenotypic analyses of lung-infiltrating immune cells, PBS-perfused lungs (left lobe) were treated with collagenase-D and DNAse1, and isolated cells were surface-immunolabelled for AM (CD45+ CD11b- CD11c+ SiglecF+), neutrophil (CD45+ CD11b+ Ly6Ghi), inflammatory monocyte (CD45+ CD11b+ Ly6chi), dendritic cell (CD45+ CD11c+ MHCII+), natural killer cell (CD45+ CD3−ve NKP46+), and T-cell markers, and analyzed by flow cytometry. Intracellular cytokine staining (ICS) of T-cells was performed as described previously.14,15 Briefly, 5 × 105 lung cells were stimulated with peptides corresponding to dominant MERS-CoV-specific CD4 (N99) and CD8 (S434) T-cell epitopes at 1 µM concentration in RPMI-10% FBS media for 6 hrs in the presence of 1 µ
Histopathology Studies
Animals were anesthetized and transcardially perfused with 10 mL PBS. Histopathology was performed on all 4 right lung lobes (i.e., cranial, middle, caudal, and accessory). Lung lobes were removed, fixed in zinc formalin (Millipore Sigma, cat # Z2902), and paraffin-embedded. Tissue sections were stained with Harris-modified hematoxylin and eosin and examined and scored by light microscopy in a blinded fashion by a board-certified veterinary pathologist (Dr. Sunil More) as described previously. 56 Control PBS and CL-liposome-treated lungs were scored for edema and fibrin with scores of 0, 1, 2, 3, and 4 representing edema and/or fibrin in 0%, less than 5%, 6%–33%, 33%–66%, and more than 66% of the examined lung lobes, respectively. Lungs were also scored for inflammatory cell infiltration, with scores of 0, 1, 2, 3, and 4 representing areas with 0%, less than 5%, 6%–33%, 33%–66%, and more than 66% of the examined lung lobes infiltrated by polymorphonuclear and/or mononuclear cell inflammation.
Statistical Analysis
Results were analyzed using Student’s t test. Data in bar graphs or bar/scatter plots are represented as mean ± standard error of the mean (SEM). Weight loss and survival curves were assessed for statistical significance using log-rank (Montel–Cox test) or Gehan–Breslow–Wilcoxon test. *P < .05, **P < .01, or ***P < .001 was used to determine statistical significance.
Results
AMs Are Protective During MERS-CoV Infection in hDPP4-KI Mice
Alveolar macrophages have a protective role in a variety of viral lung infections.29,30,51,65,77 In contrast, the role of AMs during hCoV infections, specifically during MERS-CoV infection, is not well understood. Consequently, using intranasal administration of CL-liposomes, we depleted AMs and assessed the role of AMs in MERS pathogenesis. First, we examined the depletion of AMs in naïve hDPP-KI mice following intranasal CL-liposome treatment. As shown in Fig. 1, a single intranasal administration of CL-liposome resulted in significantly reduced AM numbers in the bronchoalveolar lavage fluid of naïve mice compared to PBS controls (Fig. 1a, b). A single intranasal CL-liposome administration also caused a significant decrease in AMs in the lungs for several days post-MERS-CoV infection (Fig. 1c, d). To assess the role of AMs during MERS pathogenesis, we challenged control PBS-treated and CL-liposome-treated hDPP4-KI mice with a sublethal dose of MA MERS-CoV (clone 6-1-2, as described earlier). CL-liposome-treated hDPP4-KI mice had a significant increase in weight loss resulting in high mortality due to euthanasia, as compared to control PBS-treated hDPP4-KI mice following MERS-CoV-MA infection (Fig. 2a, b). These results demonstrate that AMs facilitate host protection during MERS-CoV infection and depletion of AMs results in severe clinical disease necessitating early euthanasia in an otherwise a sublethal infection.
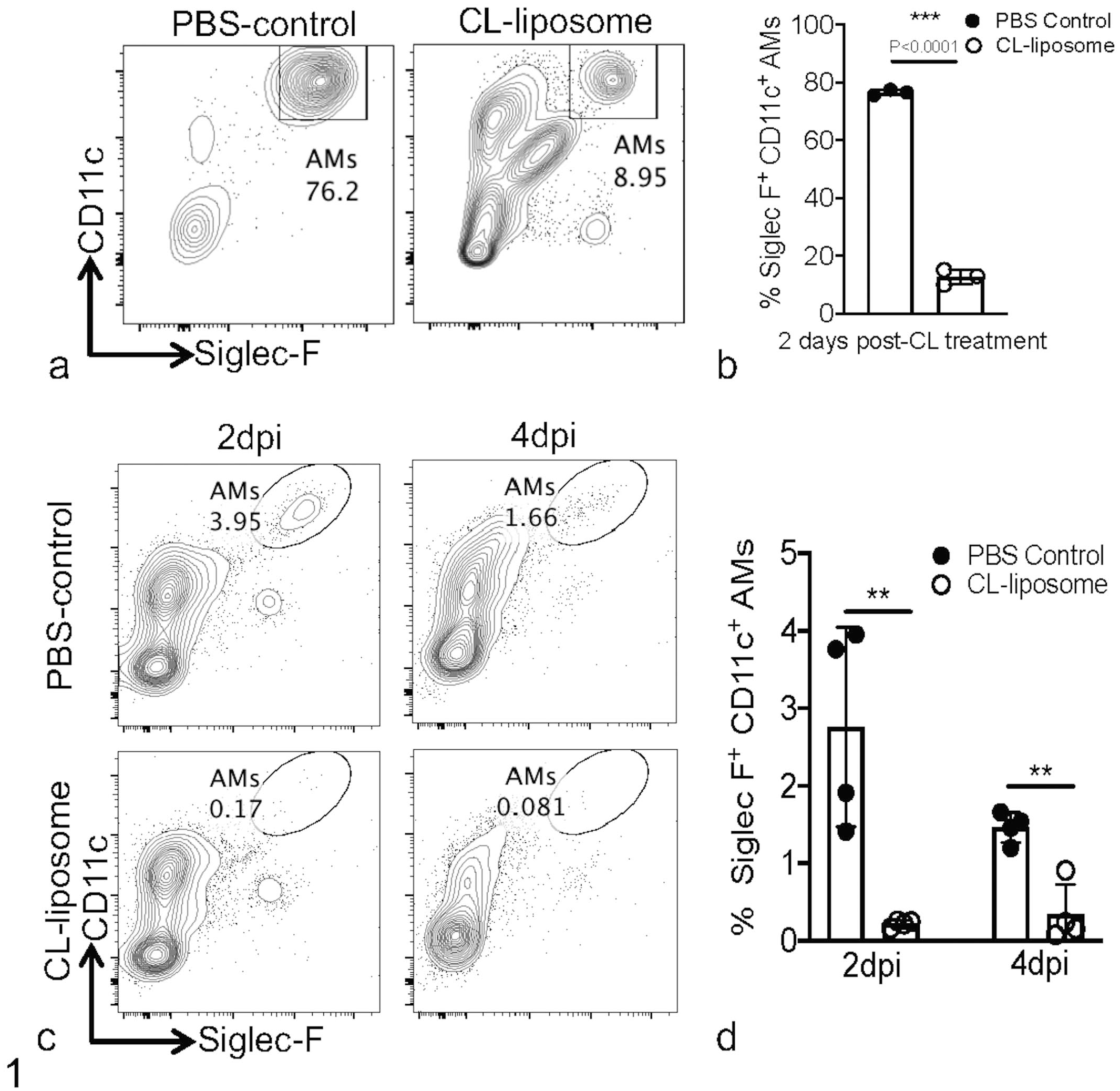
Intranasal clodronate (CL) liposome administration depletes alveolar macrophages (AMs). hDPP4-KI mice (8- to 14-week old) were treated with a single dose of 75 µL of PBS or CL-liposome 1 day before infection. (a and b) FACS plots (a) and bar graphs (b) show the percentage of AMs in the bronchoalveolar lavage fluid of PBS-treated and CL-liposome-treated mice. (c and d) FACS plots (c) and bar graphs (d) show the percentage of AMs in the lungs at 2 and 4 dpi in PBS-treated and CL-liposome-treated mice. Data are derived from one experiment (a-d) with 3-5 mice per group per experiment. Statistical significance was determined using Student’s t test with **P < .01 and ***P < .001.
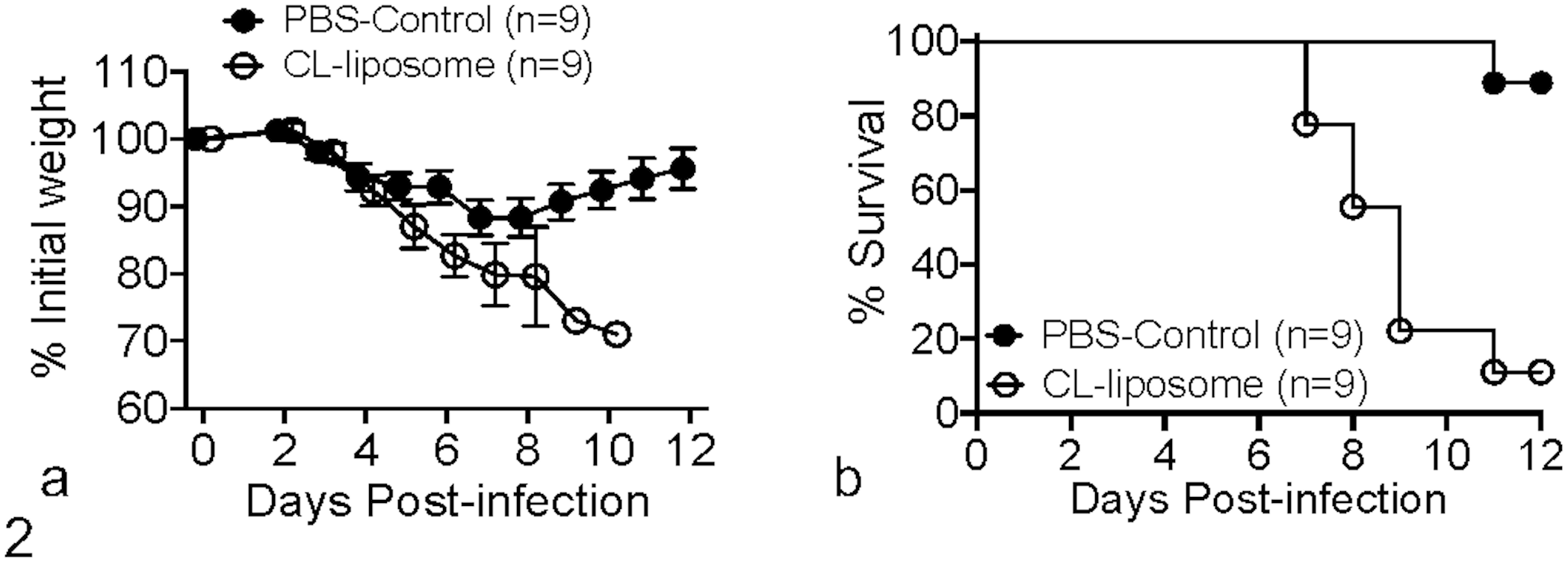
AMs protect mice from MERS-CoV-induced morbidity and mortality. hDPP4-KI mice (8- to 14-week old) were challenged intranasally with 150-300 PFU of mouse-adapted MERS-CoV and monitored for morbidity and mortality for 12 days. Weight loss (a) and survival curves (b) show morbidity and mortality in PBS-treated and CL-liposome-treated mice. (a and b) Data are pooled from 2 experiments with 3-5 mice per group per experiment.
AM Depletion Causes Differential Myeloid Cell Response During MERS
Alveolar macrophages mediate host response to virus infections by facilitating recruitment and activation of innate and adaptive immune cells. 30 To assess whether depletion of AMs alters the myeloid cell response and thereby promotes severe disease during MERS-CoV infection, control- and CL-liposome-treated mice were euthanized at early (2 dpi) and late (7 dpi) timepoints postinfection. Phosphate buffered saline-perfused lungs were analyzed for infiltration of myeloid cells such as inflammatory monocyte-macrophages (IMMs) and neutrophils. As shown in Fig. 3a to d, there was no difference in the accumulation of myeloid cells in control and Cl-liposome-treated lungs at 2 dpi. In contrast, we observed a significant increase in the percentage of IMMs and in the percentage and total number of neutrophils in the lungs of CL-liposome-treated mice at 7 dpi (Fig. 3a–d). Further analysis revealed minimal differences in other innate immune cells such as natural killer cells (data not shown). Collectively, these results indicate that AM depletion results in increased accumulation of neutrophils in the lung during later stages of infection, which may cause pathological changes and severe pneumonia.
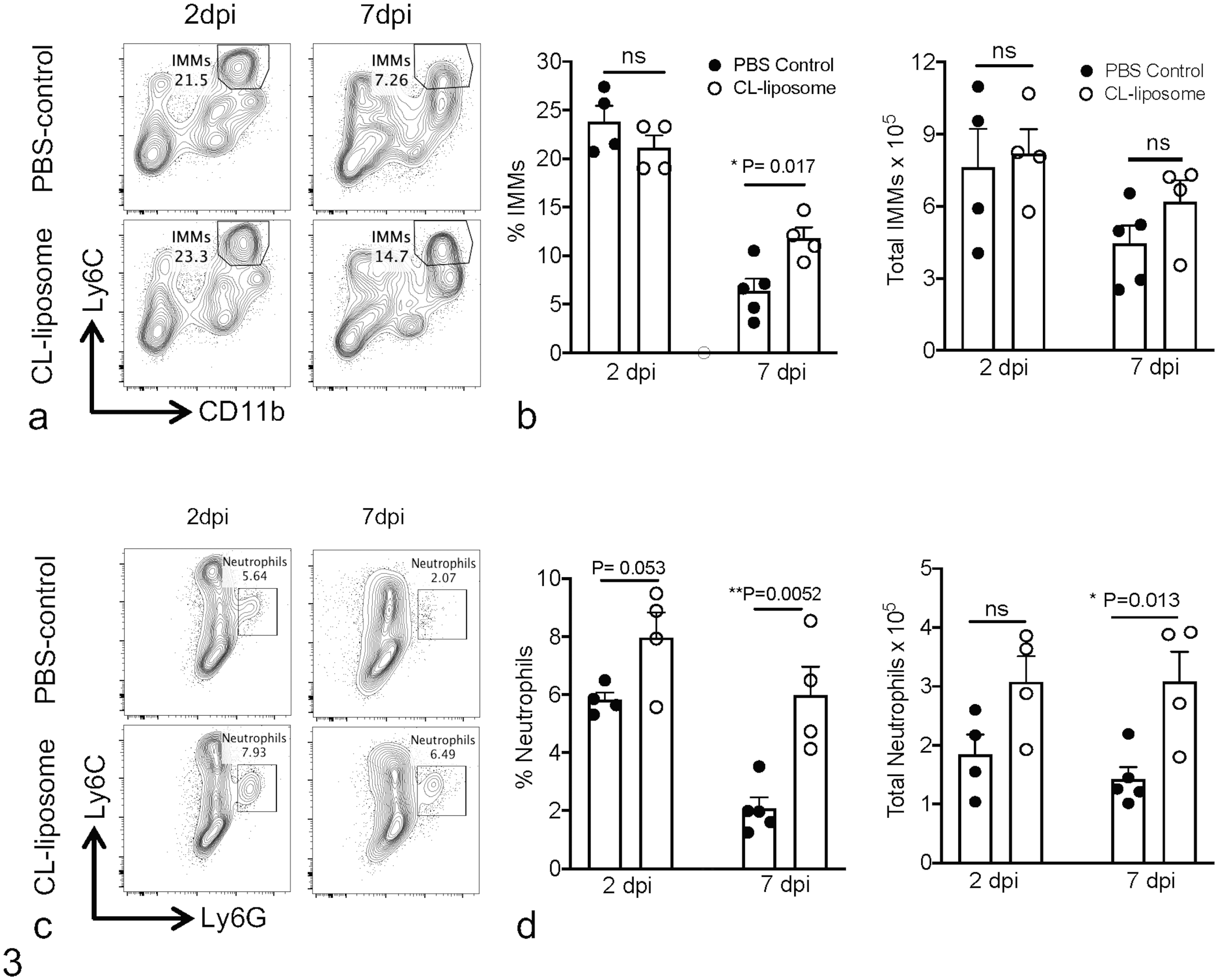
AM depletion promotes accumulation of myeloid cells in the lungs. Control PBS-treated and CL-liposome-treated hDPP4-KI MERS-CoV mice were euthanized at 2 and 7 dpi. Single-cell suspensions from PBS-perfused lungs were stained for different immune cells. (a) FACS plots show percentage of IMMs in the lungs at 2 and 7 dpi. (b) Scatter plot bar graphs show the percentage and the total number of IMMs in the lungs at 2 and 7 dpi. (c) FACS plots show the percentage of neutrophils in the lungs at 2 and 7 dpi. (d) Scatter plot bar graphs show the percentage and the total number of neutrophils in the lungs at 2 and 7 dpi. Each scatter point represents an individual mouse with 4-5 mice per group per experiment. Statistical significance was determined using Student’s t test with *P < .05 and **P < .01.
Lack of AMs Impairs T-Cell Response During MERS-CoV Infection
We observed significant morbidity and mortality in CL-liposome-treated mice beginning 5 days post-infection. Consequently, we examined whether increased mortality in AM-depleted mice correlated with a reduced MERS-CoV-specific T-cell response. Here, control PBS-treated and CL-liposome-treated MERS-CoV-infected mice were euthanized at 7 dpi, and PBS-perfused lung cells were stimulated with peptides corresponding to dominant MERS-CoV-specific CD4 and CD8 T-cell epitopes.15,82 Peptide re-stimulated T-cells were immunolabeled for interferon gamma (IFN-γ) and the percentage and the total numbers of N99-epitope-specific CD4- and S434-specific CD8-T-cells were enumerated. Our data showed marginal differences in percentages of MERS-CoV-specific CD4 and CD8 T- cells in control and CL-liposome-treated lungs (Fig. 4a–d), while the total number of IFN-γ expressing CD4 T-cells was significantly reduced in CL-liposome-treated mice (Fig. 4b). Overall, these results show that depletion of AMs impairs virus-specific CD4 T-cell response during MERS-CoV infection.
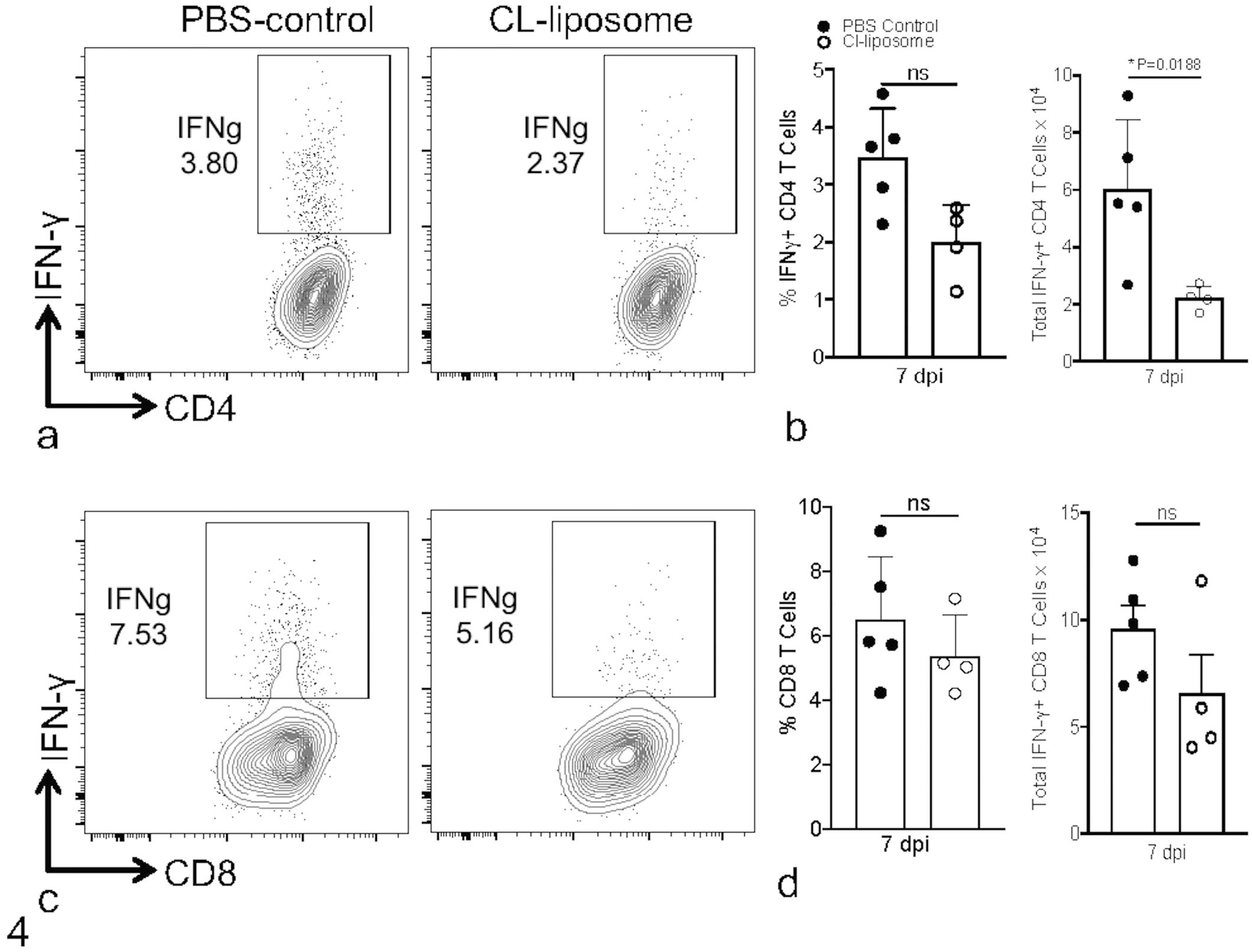
AM depletion impairs T-cell response to MERS-CoV infection. Control PBS-treated and CL-liposome-treated hDPP4-KI MERS-CoV mice were euthanized at 7 dpi. Single-cell suspensions from PBS-perfused lungs were stained for different immune cells. (a and c) FACS plots show the percentage of CD4+ (a) and CD8+ (b) T-cells in the lungs at 7 dpi. (b and d) Scatter plot bar graphs show percentage and the total number of CD4+ (b) and CD8+ (d) T-cells in the lungs at 7 dpi. Each scatter point represents an individual mouse. Statistical significance was determined using Student’s t-test with *P < .05.
AMs Protect Mice From MERS-CoV-Induced Lung Inflammation
To assess lung lesions in PBS-treated and CL-liposome-treated mice, we examined right cranial, middle, caudal, and accessory lungs lobes collected from both groups of mice at 2, 4, and 7 dpi. Our data showed reduced edema/fibrin deposition in CL-liposome-treated lungs, and no difference in lung inflammatory infiltrates in control and CL-liposome-treated mice at 2 dpi (Figs. 5–7). However, a significant increase in alveolar and airway edema and fibrin deposition was observed in CL-liposome-treated lungs, despite a similar lung inflammatory cell infiltrate score in both groups of mice at 4 dpi (Figs. 5–7). Furthermore, alveolar edema and fibrin deposition were comparable in the lungs of both groups of mice at 7 dpi, while increased lung inflammatory cell infiltration, as characterized by the area of mono- and polymorphonuclear cell accumulation in the alveolar and interstitial space, was observed in CL-liposome-treated compared to PBS-treated control mice (Figs. 5–7). These results collectively demonstrate that AM depletion promotes lung edema, fibrin, and inflammatory cell accumulation during later stages of infection, when increased weight loss and mortality are observed in CL-liposome-treated mice.
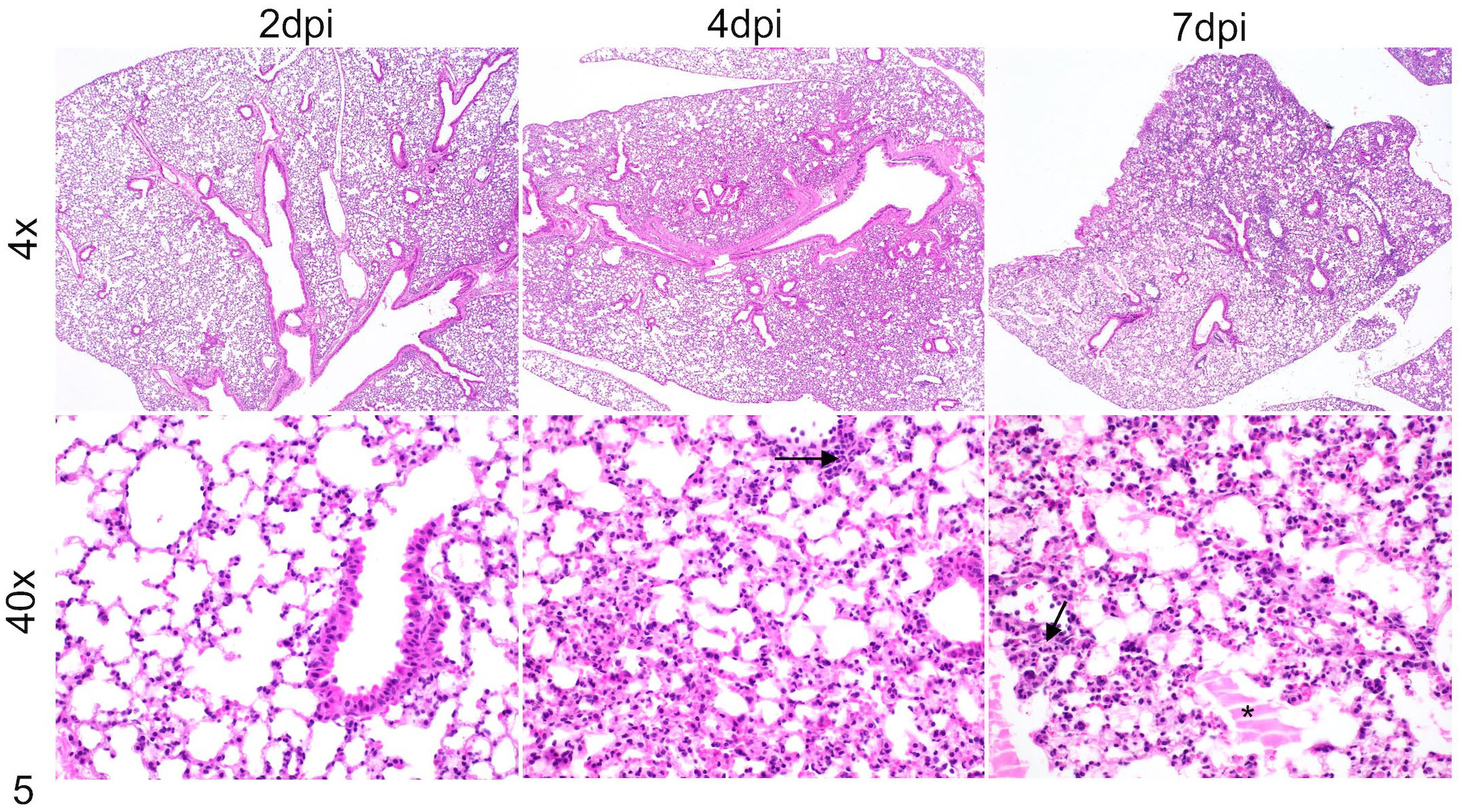
Edema, fibrin, and inflammatory cells in the lungs of control MERS-CoV-infected mice. MERS-CoV-infected control PBS-treated hDPP4-KI mice were euthanized at 2, 4, and 7 dpi. PBS-perfused lungs collected in Zn-formalin were stained with hematoxylin and eosin to assess lung lesions. Representative photomicrographs show edema and fibrin accumulation (* asterisk) and inflammatory cell infiltration (arrows) in the lungs.
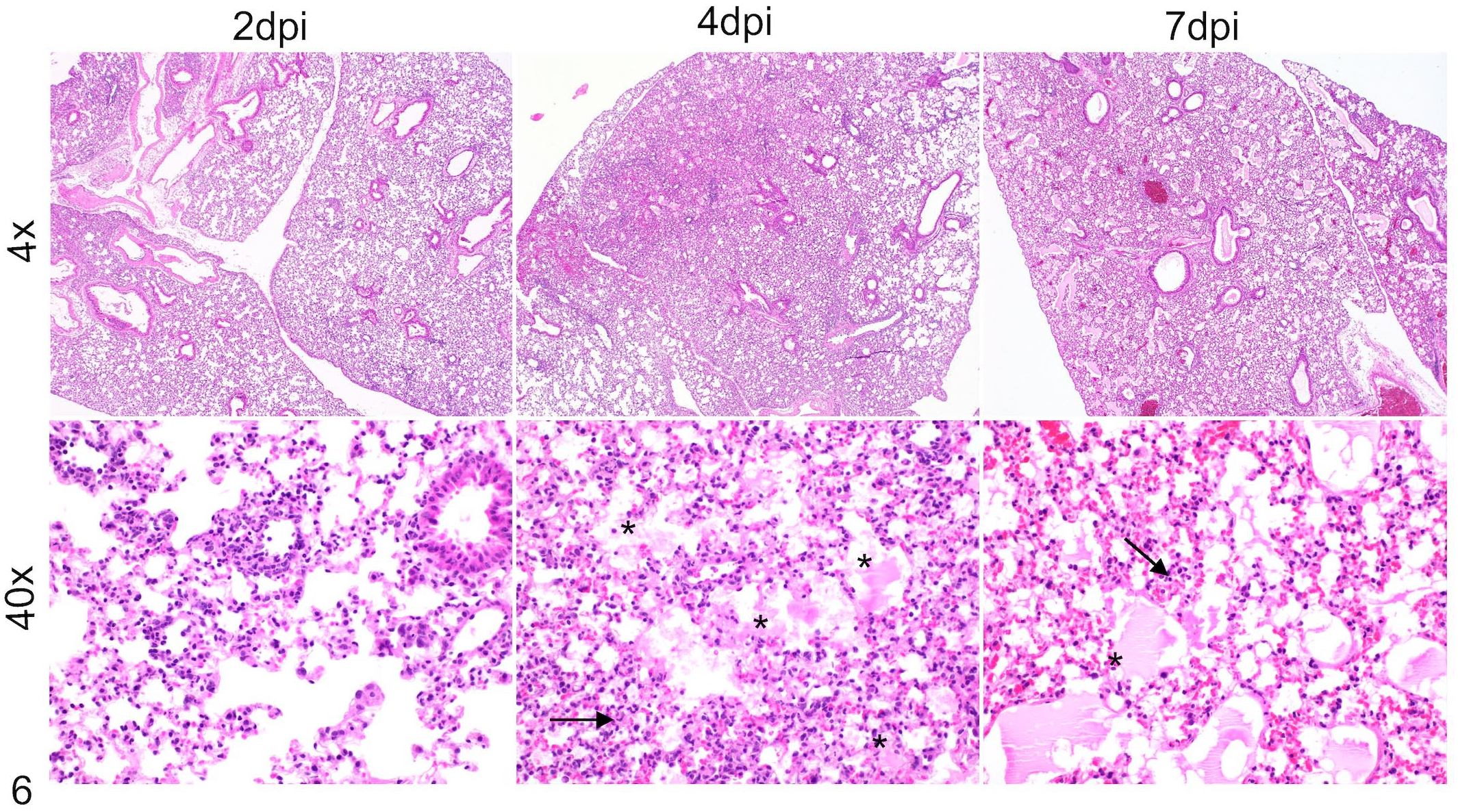
Edema, fibrin, and inflammatory cells in the lungs of AM-depleted MERS-CoV infected mice. MERS-CoV-infected CL-liposome-treated hDPP4-KI mice were euthanized at 2, 4, and 7 dpi. PBS-perfused lungs collected in Zn-formalin were stained with hematoxylin and eosin to assess lung lesions. Representative photomicrographs show edema and fibrin accumulation (* asterisks) and inflammatory cell infiltration (arrows) in the lungs.
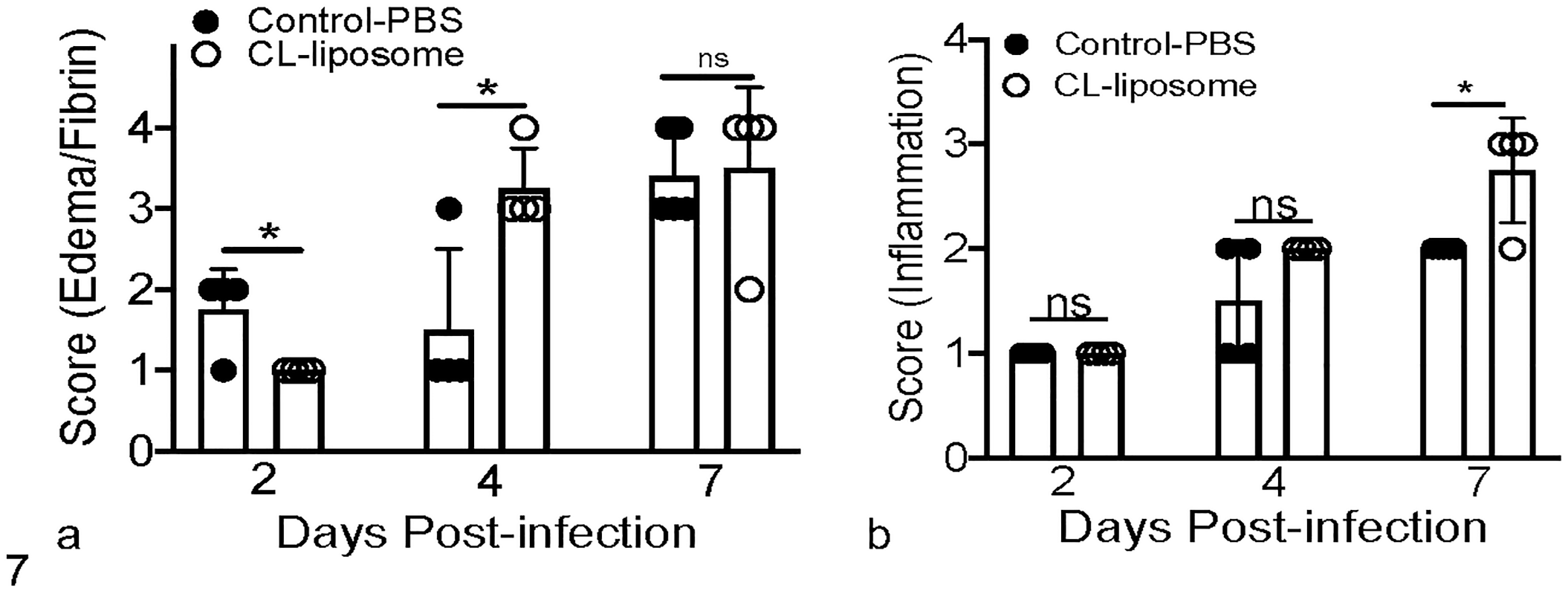
Histologic scoring of edema/fibrin and inflammatory infiltrates. Scatter plot bar graphs show average edema/fibrin (a) and inflammatory infiltrate (b) scores in lungs of control PBS-treated and CL-liposome-treated MERS-CoV-infected mice at 2, 4, and 7 dpi. Each scatter plot point represents an individual mouse. Statistical significance was determined using Student’s t test with *P < .05.
Discussion
Alveolar macrophages play a protective role following infection with pneumotropic viruses such as influenza A virus and respiratory syncytial virus.30,35,51,65,87 Recent clinical studies in SARS-CoV-2-infected individuals demonstrate that AM dysfunction is associated with elevated inflammatory cytokine/chemokine levels and severe COVID-19. 75 Conversely, studies in animal models show a pathogenic role for AMs following infection with several pneumotropic viruses such as human metapneumovirus, 48 mouse hepatitis virus 1 (MHV-1), 34 and SARS-CoV. 86 However, the role of AMs in the pathogenesis of MERS-CoV infection is not well understood. Therefore, using an intranasal CL-liposome-mediated AM depletion strategy, we evaluated the role of AMs during MERS-CoV infection in mice.
Our results demonstrate that AM depletion increases morbidity and mortality during MERS-CoV infection. Alveolar macrophages are a subset of lung resident macrophages that populate lungs during early development and are one of the first immune cells that respond to a pathogen challenge in the lower airways.25,30,33,65,77 Alveolar macrophages are either productively or abortively infected with pneumotropic viruses,24,25,29,30 sense viral nucleic acids via RIG-I/MDA5/MAVS signaling, 51 and induce an antiviral IFN/ISG response to counter virus infections.25,30,51,77 Similarly, while AMs infected with hCoV-229E or SARS-CoV-2 harbor significantly elevated amounts of viral genomic RNAs, no infectious virus is detected in AM cultures.24,29 In further comparison to influenza A virus and respiratory syncytial virus infections, which induce a robust IFN response, SARS-CoV and SARS-CoV-2 fail to induce IFN responses in AMs and are thus likely evade the host antiviral response.24,29 Of note, ultrastructural examination of lungs from MERS patients showed viral particles in AMs. 5 However, whether AMs phagocytose the virus or MERS-CoV abortively infects AMs and thus induces a detectable antiviral IFN/ISG response in the lungs requires further investigation.
Alveolar macrophage depletion using CL-liposomes leads to a reduced MERS-CoV-specific CD4 T-cell response and increased morbidity and mortality in hDPP4-KI mice infected with a sublethal dose of MERS-CoV. These results contrast with our SARS-CoV studies showing that AMs promoted viral evasion of host immunity by suppressing lung dendritic cell migration to draining lymph nodes, leading to a reduced SARS-CoV-specific T-cell response. 86 In these studies, depletion of AMs facilitated a robust migration of CD103+ lung dendritic cells to draining lymph nodes, increased virus-specific T-cell responses in the lungs, and thus provided significant protection against lethal SARS. 86 Robust AM-mediated T-cell responses, on the contrary, are also shown to cause immune-mediated damage during MHV-1 infection. 34 Similarly, a recent COVID-19 clinical study provided correlative evidence for the role of AMs in causing T-cell-mediated persistent lung disease. 31 Although we show reduced MERS-CoV-specific CD4 T-cells in AM-depleted lungs, it is unclear from our studies whether the suboptimal CD4 T-cell response is due to (1) suppression of T-cell responses by neutrophils and myeloid-derived suppressor cells 64 or (2) depletion of a subset of dendritic cells and thus impaired T-cell priming in CL-liposome-treated mice. 20
In addition to innate IFN/ISG and adaptive T-cell-mediated antiviral responses, AMs help to protect the host by (1) preventing virus infection of type-I alveolar epithelial cells,25,30,51 (2) limiting lung inflammation by suppressing tissue-resident macrophage-mediated inflammation through peroxisome proliferator-activated receptor (PAPRγ) activation, 39 and (3) promoting tissue tolerance via forkhead box protein P3 (Foxp3) induction in naïve T-cells. 22 Alveolar macrophages also regulate the recruitment and activation of myeloid cells such as neutrophils. 11 In agreement with the latter results, we observed a significantly increased accumulation of neutrophils in AM-depleted lungs. Such an increase is likely due to elevated production of myeloid cell chemoattractants in the absence of AMs. 11 In addition, T-cells moderate an overactive innate immune response caused by mimics of viral PAMPs, endotoxins, and inflammasomes by producing interleukin (IL)-10 and other immunomodulatory molecules.23,32,41,42,46,71,72 Recent studies from our and other laboratories show that hCoV-specific T-cells are required for virus clearance and host protection.18,83–86 Consequently, reduced CD4 T-cell responses may result in impaired virus clearance and/or reduced host tolerance leading to robust neutrophil accumulation, uncontrolled lung inflammation, and fatal pneumonia.
Our results also show a correlation between FACS studies and histopathology findings as demonstrated by an increased percentage of myeloid cells (neutrophils and monocyte-macrophages) and increased lung inflammatory cell inflammation score in CL-treated lungs at 7 dpi. Our recent studies showed that robust myeloid cell activity caused severe disease, with minimal effect on virus titers.14–16 Therefore, it is likely that robust myeloid cell-induced inflammation plays a key role in causing severe pneumonia in AM-depleted mice. Furthermore, increased edema in CL-treated lungs at 4 dpi is likely due to lack of AMs, which are shown to play an important role in alveolar edema clearance. 63 Limited lung pathology data from MERS patients and several autopsy studies from the SARS epidemic showed diffuse alveolar damage, edema, and fibrin deposition associated with robust inflammatory cell infiltration.6,28,40,50,58 Recent studies that used murine models to study MERS-CoV infection also showed several histopathology features including diffuse alveolar damage as observed in humans.21,52,69 Our histopathological examination of lungs from control- and AM-depleted mice showed airway edema, fibrin distribution, and mono- and polymorphonuclear cell accumulation in the lungs. However, we did not observe diffuse alveolar damage, a prominent feature observed in human patients and murine models with lethal MERS. This is likely due to the sublethal dose (150-300 PFU/mouse) used in our studies compared to lethal dose of virus inoculum (2 × 104–106 PFU/mouse) and an extensive virus-induced alveolar damage in other models of lethal MERS.21,52,69 Increased airway edema and fibrin deposition observed in our study indicate lung microvascular leakage, impaired gas exchange, and hypoxia, all of which were observed in patients with fatal CoV infection.7,8,58,66,76 Since robust myeloid cell activity causes lung microvascular leakage, 14 and AMs suppress infiltration of these cells, it is possible that AMs indirectly control vascular leakage. In addition, increased myeloid cell accumulation suggest a role for myeloid cell-mediated “cytokine storm” and lung inflammation. Nevertheless, several histopathology features in AM-depleted lungs correlate with microscopic features observed in the lungs of patients with lethal SARS and MERS.
Overall, our results show that depletion of AMs increases morbidity and mortality in hDPP4-KI mice infected with an otherwise sublethal dose of MERS-CoV-MA. Although the effect of AM depletion on lung MERS-CoV titers is not known, we speculate based on our earlier studies14–16 that increased myeloid cell response or reduced virus-specific CD4 T-cell responses or both facilitate severe MERS in AM-depleted mice. Consequently, elucidating the molecular mechanisms that regulate AM-mediated myeloid cell accumulation and T-cell responses and their effect on virus titers will be critical to better understanding the pathogenesis of MERS-CoV and other hCoVs infections.
Footnotes
Author Contributions
RC and SP conceptualized the study; RC and MS carried out experiments and data analysis; SM carried out histopathology evaluation; RC and SP wrote the manuscript; RC, MS, SM, and SP reviewed and edited the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported in part by an institutional research fund to RC from Oklahoma State University, College of Veterinary Medicine, Oklahoma Center for Respiratory and Infectious Disease (OCRID) Centers of Biomedical Research Excellence (COBRE) grant NIH P20GM103648 and NIH AG060222. Part of this work is also supported by NIH P01 AI060699 (SP) and RO1 AI129269 (SP). We also thank OCRID/OSU Immunopathology core and Mr. Curtis Andrews for the preparation of histopathology slides.