
Abstract
Classification of pneumonia in animals has been controversial, and the most problematic pattern is interstitial pneumonia. This is true from the gross and histologic perspectives, and also from a mechanistic point of view. Multiple infectious and noninfectious diseases are associated with interstitial pneumonia, all of them converging in the release of inflammatory mediators that generate local damage and attract inflammatory cells that inevitably trigger a second wave of damage. Diffuse alveolar damage is one of the more frequently identified histologic types of interstitial pneumonia and involves injury to alveolar epithelial and/or endothelial cells, with 3 distinct stages. The first is the “exudative” stage, with alveolar edema and hyaline membranes. The second is the “proliferative” stage, with hyperplasia and reactive atypia of type II pneumocytes, infiltration of lymphocytes, plasma cells, and macrophages in the interstitium and early proliferation of fibroblasts. These stages are reversible and often nonfatal. If damage persists, there is a third “fibrosing” stage, characterized by fibrosis of the interstitium due to proliferation of fibroblasts/myofibroblasts, persistence of type II pneumocytes, segments of squamous metaplasia of alveolar epithelium, plus inflammation. Understanding the lesion patterns associated with interstitial pneumonias, their causes, and the underlying mechanisms aid in accurate diagnosis that involves an interdisciplinary collaborative approach involving pathologists, clinicians, and radiologists.
Keywords
The diagnosis of interstitial pneumonias (IPs) in animals has not been fully elucidated and frequently carries problems at the moment of identification, not only from a pathologic perspective, but also from a mechanistic point of view. This review defines and differentiates the terms frequently used in veterinary lung pathology, discusses the mechanism of diffuse alveolar damage (DAD), and then discusses the many different causes of this condition. This approach allows a comprehensive understanding of this interesting and important disease pattern.
The presence of an intact endothelial-epithelial barrier is essential for effective pulmonary gas exchange. Pulmonary insults (eg, pneumonia, aspiration, lung trauma) and nonpulmonary insults (eg, sepsis) can all injure this barrier, leading to hypoxemia as a result of edema and inflammation of the alveolus. The morphology of the alveolus and the alveolar wall has been extensively studied and is mainly composed of 5 different types of cells: type I pneumocytes mediate gas exchange, type II pneumocytes produce surfactant and have the ability to proliferate and also perform immune functions, endothelial cells have gas exchange and metabolic functions, alveolar macrophages coordinate host defenses, and interstitial fibroblasts provide alveolar structure through the production of extracellular matrix. 70 In horses and ruminants, pulmonary intravascular macrophages (PIMs) also play a role in pulmonary inflammation, because they are able to stimulate acute lung inflammation by recruiting inflammatory cells. 153 This review addresses the most important interactions among these cells, the result of these interactions, and the progression of lung damage, focusing on etiologies that affect pneumocytes and endothelial cells.
Classification of pneumonia has always been controversial. Approaches to this issue have varied over the years, with different authors basing their approach on different parameters. Strategies can be based on cause (bacterial, viral, fungal, parasitic), type of exudation (suppurative, fibrinous, pyogranulomatous), epidemiologic attributes (eg, “enzootic pneumonia,” “shipping fever”), anatomic distribution (cranioventral, diffuse, multifocal), etiologic agents (eg, pasteurellosis, bordetellosis), and other less commonly used classification methods. This variability is highly confusing to both students and pathologists. 103 This review uses the anatomic distribution of lesions to classify the gross presentation of pneumonia. Gross presentations are classified as suppurative bronchopneumonia, fibrinous bronchopneumonia, embolic pneumonia, granulomatous pneumonia, or IP.12,13,24,103
IP has the most complicated gross presentation and interpretation. Animals that are affected by this pattern of lung damage frequently have pulmonary parenchyma that is totally compromised, with little or no normal tissue remaining. This diffuse involvement of the lung can lead to a failure to make an accurate gross interpretation or to miss this diagnosis (Paul Stromberg, personal communication). Lungs affected with IP usually fail to collapse and are diffusely wet, heavy, meaty, slightly firm, and have prominent rib impressions.26,103 Other diffuse pulmonary processes, such as generalized edema, can often be misinterpreted as IP at the gross perspective, adding one more dimension of complexity to this already complicated pattern.
These complexities are only the “tip of the iceberg” regarding this pattern. There are several gray areas in the histologic interpretation and morphologic diagnosis of this lesion, which frequently overlaps with other pulmonary patterns. With this in mind, a beginning point is to define and discuss several terms that are found in the veterinary literature.
Interstitial lung disease (ILD) is the morphologic term currently used in veterinary medicine to describe damage involving the alveolus or interlobular septa, which form the interstitium of the lung. 26 The term ILD can sometimes be misleading, because a large array of pulmonary disorders involve damage to the distal pulmonary parenchyma, including airways, alveoli, blood vessels, and/or pleura. 138 The initial injury to the pulmonary parenchyma can involve the hematogenous route (eg, drugs, immune disease) or be airborne (minerals, fibers, dust). In response, resident cells recruit and activate inflammatory cells, which can lead to chronic inflammation and fibrosis. 138 In veterinary medicine, ILD defines a broad range of conditions including granulomatous and eosinophilic pneumonia, mononuclear infiltrates in the interstitium (such as in cases of porcine respiratory and reproductive syndrome [PRRS] or small ruminant lentivirus), pneumoconiosis (silicosis, asbestosis), lesions dominated by necrosis (such as Toxoplasma gondii infection), lesions dominated by epithelial proliferation and fibrosis (such as equine multinodular pulmonary fibrosis, caused by equine herpesvirus-5), and DAD. 26 It has been estimated that ILD accounts for approximately 20% of all lung disease in humans. 119 Regretfully, in veterinary medicine, cases of ILD are poorly characterized and relatively infrequently diagnosed, mostly because an accurate classification of ILD requires multidisciplinary collaboration between clinicians, radiologists, and pathologists. Unfortunately this collaborative approach is rarely taken.80,138 From the radiologist perspective, the protocols for acquisition of images and the terminology used to describe lesions lack uniformity. 109 From the pathologist point of view, when the interstitium is thickened, the term ILD can be used very broadly and is frequently used as an “one-size-fits-all” descriptor, independent of the underlying mechanism. Several different causes for ILD cause a similar morphologic pattern, which makes it very difficult to correlate gross and histologic findings with the cause. When the ILD histologic pattern is present in a tissue section, a long list of causes come to mind, accompanied by a knot in the stomach. The pathologist, often provided with minimal clinical information, wonders if a specific cause will be identified or if it will be another morphologic diagnosis with multiple potential causes.
In veterinary medicine, there is much confusion when using the term IP. It has been historically used as synonym for several different entities: the clinical syndrome interstitial pneumonitis, acute lung injury/acute respiratory distress syndrome (ALI/ARDS), ILD from a pathogenic/pathologic perspective, DAD, and depending on the species, for a certain group of conditions (eg, fog fever or atypical IP), with or without pulmonary fibrosis. As a general rule, it is used to describe a broad range of infectious (eg, viral) and noninfectious (eg, toxic) diseases of the interstitium, or to describe increased numbers of leukocytes in alveolar septa (such as in PRRS). It can be used every time there is damage to the alveolar epithelium, capillaries (endothelium), or increased cellularity in the interstitium. To consider only increased cellularity of the interstitium, 2 good examples of infectious causes are frequently observed, such as with PRRS and small ruminant lentivirus. In the case of PRRS, the virus has a selective tropism to monocytes, infecting first those of the lung and upper respiratory tract with subsequent viremia, spreading to macrophages throughout the body.65,104,144 Lung lesions are characterized by septal thickening by macrophages, alveolar necrotic debris and macrophages, occasional syncytial cells, mild type II pneumocyte hyperplasia, perivascular infiltrates of lymphocytes and plasma cells, and hypertrophy of peribronchiolar lymphoid tissue. 143 With small ruminant lentiviruses, the virus targets monocytes as well, which act as a Trojan horse to spread the virus through the body. 136 The histopathological finding is diffuse accumulation of lymphocytes, plasma cells, and macrophages in the lung parenchyma, plus the presence of hyperplastic lymphoid follicles around vessels and airways.26,120 Mild fibrosis and hyperplasia of smooth muscle cells are observed in the lung, with eventual destruction of tissue architecture and function. 15 In cases of small ruminant lentivirus, hyperplasia of type II pneumocytes is prominent in goats infected with caprine arthritis encephalitis virus and mild in cases of maedi. 26
Next to consider are agents that induce direct damage to alveolar epithelial cells or endothelial cells, which characterizes DAD.
ARDS and DAD
Two initial concepts need to be clarified: DAD and ARDS. DAD was proposed in 1967 to describe a type of lung injury characterized by “endothelial and alveolar lining cell injury which leads to fluid and cellular exudation and in some cases progresses to extensive interstitial fibrosis.” 81 In contrast, ARDS is a clinical definition that refers to a form of hypoxemic respiratory failure characterized by severe impairment in gas exchange and lung mechanics with a high case fatality rate. 66 It is important to differentiate that DAD is a histologic pattern with known progressive stages, and ARDS is a clinical definition. ARDS and DAD should not be used interchangeably.
Understanding this difference allows the 2 terms to be used in the same sentence. DAD is the most frequent (approximately 50%) histologic finding in humans and animals with ARDS.21,25,113,147 In humans, ARDS characterizes critically ill adults and children with acute hypoxemia, noncardiogenic pulmonary edema, reduced lung compliance (increased “lung stiffness”), increased work of breathing, and the need for positive pressure ventilation. It is associated with several different clinical disorders, such as trauma, pneumonia, sepsis, and aspiration. 5 Clinically, it is defined by an acute onset, defined as occurring within 1 week of a known clinical insult or new/worsening respiratory signs, bilateral chest radiographic infiltrates that are not fully explained by effusions, lobar/lung collapse or nodules, impaired oxygenation (defined as PaO2/FiO2 <300 mm Hg value positive end-expiratory pressure of at least 5 cm H2O), and either a pulmonary artery occlusion pressure of ≤18 mm Hg or no evidence of left atrial hypertension.10,49,113 This definition only includes clinical variables. Other characteristics relating to histopathology or pathobiology of the process were not included because these variables were not considered clinically feasible. 49 Development of ARDS is more frequent in older patients, which is explained by increased susceptibility to pneumonia and sepsis. 79 Other causes for ARDS that would explain the remaining nearly 50% of the human cases in which DAD is not observed include infections (viral, fungal, and bacterial including leptospirosis), pulmonary hemorrhages, acute eosinophilic pneumonia, drugs, pulmonary mycotoxicosis, pulmonary edema, and pulmonary embolism.21,45,147 Many cases of pneumonia can have features of DAD without necessarily fitting the criteria for ARDS, and contrarily, the presence of DAD is not the only lesion that can result in ARDS. This is particularly important, as patients who meet the clinical definition of ARDS and have DAD appear to have worse outcomes than patients with ARDS but without DAD. 21
Human ARDS has been carefully analyzed and compared with the animal scenario, and 5 criteria (4 required and a fifth highly recommended) for the diagnosis of this condition were established and named VetARDS.182,183 Briefly, VetARDS includes acute onset of dyspnea of less than 72 hours duration, identifiable risk factors, pulmonary capillary leakage without evidence of a cardiogenic origin, evidence of inefficient gas exchange, and evidence of diffuse pulmonary inflammation. The details for physiologic parameters and testing required for VetARDS can be found elsewhere. 182 Clinically, VetARDS is characterized by rapid onset of respiratory distress, increased respiratory rate and heart rate, increased respiratory effort with decreased chest excursion and prominent abdominal component or paradoxical breathing patterns, and hypoxemia minimally responsive to oxygen supplementation with hypocapnia, normocapnia, or hypercapnia. 41 Three definitions were proposed: VetARDS, a syndrome that resembles ARDS in human medicine; NERDS (neonatal equine respiratory distress syndrome), a syndrome that occurs during the first 24 hours after birth; and EqNARDS, described in foals less than 1 week of age, which is similar to VetARDS except that an age-specific progressive scale of the PaO2/FiO2 ratio is used to document hypoxemia.41,182
ARDS is a clinical definition, whereas DAD is the hallmark histopathologic finding of ARDS with specific histologic features. ALI is defined as a milder form of ARDS. 77 Currently, ALI/ARDS can be considered as mild (PaO2/FiO2 of 201–300 mm Hg), moderate (PaO2/FiO2 of 101–200 mm Hg), and severe (PaO2/FiO2 of ≤100 mm Hg).
Diffuse Alveolar Damage
DAD is a type of lung injury characterized by an orderly sequence of pathologic changes after lung injury. The stages of DAD are divided into 3 overlapping phases: an acute or exudative phase, an organizing subacute phase, and a fibrotic or chronic phase. Once DAD is triggered, the exudative and organizing phases will occur, and in most cases, the lung will recover its function. DAD will not always evolve to the fibrotic phase. If the stimulus is removed and basement membranes are intact, normal architecture is reestablished.3,25,87,160
The main histologic features for each stage are summarized in Table 1. The different stages of DAD and its associated molecular mechanisms are elegantly reviewed elsewhere. 113
Summary of microscopic findings in the different stages of diffuse alveolar damage.

Exudative Phase
The first stage is the exudative (acute) phase, which is characterized by damage to the internal lining of alveoli (either type I or II pneumocytes) or damage to endothelial cells. 174 Type I pneumocytes are more sensitive to injury than type II pneumocytes. 70 After the inciting injury, there are several physiologic features of the alveolar space that gets disrupted. From a physiologic perspective, a small amount of fluid can be found in the normal alveolar space, which is tightly regulated primarily by apical Na+ channels, and basolateral Na+ and K+ ATPase pumps within the type I and II pneumocytes.111,112 Consequently, damage to the alveolar epithelial cells or direct damage to endothelial cells results in either lack of regulation of this feature or alveolar flooding, resulting in alveolar edema. 111 Resident alveolar macrophages do not perform well in fluid environments, and their function and ability to deal with foreign particles get disrupted by this edema fluid. At the same time, and depending on the inciting stimulus, resident alveolar macrophages and epithelial cells can release cytokines, such as tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, IL-6, and IL-8115,117,174 and chemokines, including CCL2, CXCL8, CXCL1, and CCL20 158 for the recruitment of neutrophils, which will release numerous proteases, reactive oxygen species (ROS) and neutrophil extracellular traps (NETs).129,179 This increases cell-mediated and non–cell-mediated damage to cells within alveoli. Inflammatory macrophages and neutrophils can also release factors to induce apoptosis of epithelial cells (such as FasL and TRAIL), which will create loss of tight junctions between epithelial cells.69,73 This is also triggered by the activation of inflammation that allows the arrival of inflammatory cells and also induces several vascular changes. As a result, alveoli are flooded and hyaline membranes are present along the denuded alveolar surface.
Primary damage directed to endothelial cells can result in altered endothelial physiology and barrier function and an increased permeability syndrome which will overload the capacity of the interstitial lymphatics to drain fluid. 77 Endothelial cells release increased amounts of von Willebrand factor (vWf) and upregulate the intracellular adhesion molecule-1 (ICAM-1).19,115,173 Moreover, endothelial cells are able to activate and amplify an inflammatory stimulus. 113 Inflammatory cells, particularly neutrophils, make their way through the vascular wall, leading to basement membrane destruction and increased permeability of the alveolar capillary barrier.77,174,189 In this stage, the most significant histologic findings are alveolar edema, occasional alveolar hemorrhages, absent or mild neutrophilic infiltration, and the presence of alveolar foamy macrophages. The key finding that defines DAD is hyaline membranes, which are composed of cellular debris, plasma proteins (albumin, fibrinogen, and immunoglobulins) and surfactant components (Fig. 1).56,160 Immunohistochemical studies of the hyaline membranes reveal that these structures can be highly heterogeneous in their components, which can be the result of different formation mechanisms. The main components of hyaline membranes are derived from alveolar epithelial cells and their proteins and also serum factors, but fibrin was not a frequent finding. 156 In contrast, cases of human COVID-19 with DAD have been characterized by hyaline membranes formed by the polymerization of fibrin following leakage of plasma fluid into the alveolar spaces. 8 This finding further supports the heterogeneity that exists in composition of hyaline membranes.
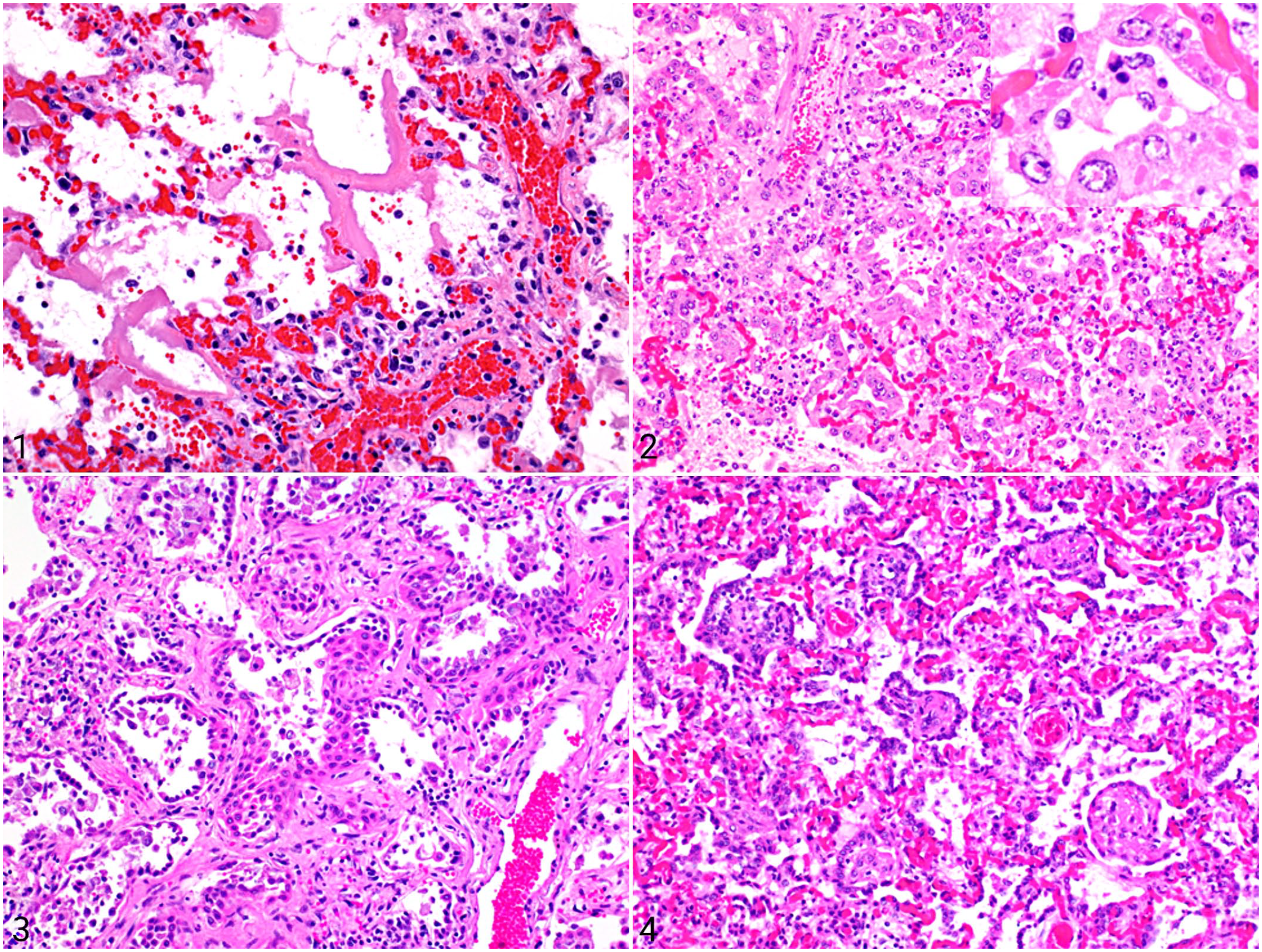
Microscopic findings in the lung of animals with diffuse alveolar damage (DAD). Hematoxylin and eosin.
Fibrin thrombi in various stages of organization are often present in small pulmonary vessels and they may be numerous. Capillary thrombosis is associated with endothelial dysfunction–associated upregulation and activation of tissue factor and damage to the ability to activate the vitamin K–dependent proteins C and S. This local procoagulant state potentiates pulmonary dysfunction and the acute inflammatory response. 7 In addition to activation of endothelial cells, the role of vascular megakaryocytes is currently recognized as a determinant for prothrombotic status leading to platelet activation, activation, and consumption.106,167
This means that any time there is a primary lesion to any of these structures (pneumocytes or endothelial cells), direct injury to the alveolus may be exacerbated with a secondary wave of inflammatory injury. 113 If the lesion is very recent (less than 24 hours), the only histologic finding in alveoli is edema, few fibrin strands and hemorrhages, without a strong cellular component. For this reason, the clinical history is a very important factor to consider. The diagnosis can be elusive if all factors are not included.
Proliferative Phase
Patients surviving the exudative phase enter the proliferative or organizing (subacute) phase, which appears 3 to 6 days after initiation of the alveolar injury. Once type I pneumocytes are injured, adjacent type II pneumocytes are stimulated to multiply and transdifferentiate into type I pneumocytes, so the key finding for this stage is type II pneumocyte hyperplasia (Fig. 2), which is frequently characterized by highly reactive cuboidal to columnar cells that upholster the alveolar surface.26,103,129 There are reports that type II pneumocytes can arise from cells other than themselves, including surfactant-protein-expressing embryologic precursors, club cells, and bronchoalveolar stem cells located at the bronchoalveolar junction and also various cells derived from the circulation.31,85,89,129 Regenerative epithelial cells show a variable degree of atypia consisting of nuclear enlargement, clumped nuclear chromatin, large eosinophilic nucleoli, and frequent mitoses.25,160 The cytologic atypia in proliferating pneumocytes can be very pronounced and may be a source of false-positive cytology for a neoplastic process in bronchoalveolar lavage specimens. 11 This hyperplasia can be mediated by different factors that are released from other epithelial cells (such as keratinocyte growth factor, epidermal growth factor, and hepatocyte growth factor) and also from fibroblasts and activated platelets.38,113 The final purpose of this stage is to cover the denuded surface of the alveolus with type II pneumocytes, which will finally differentiate to type I pneumocytes. The molecular process by which this transformation is achieved is not completely understood 129 It has been demonstrated that NK homeobox 2-1 (Nkx2-1) expression in type I pneumocytes is necessary for their development and maintenance and is now recognized as the first overarching factor in the transcriptional hierarchy in the biology of these cells. 100 The activity of the water pumps is restored and with that alveolar macrophages are able to remove cellular and noncellular debris in the alveolus. At this stage, there is also initial proliferation of fibroblasts and myofibroblasts in the alveolar interstitium, with the initial deposition of collagen. These combine to thicken the alveolar septa. 56 Recent research has shown that neutrophils actually have a large role in this stage by releasing lipid mediators and blocking chemotaxis, releasing metalloproteinases that will have a fundamental role in chemokine cleavage, production of NETs for chemokine trapping, and also migrating into the alveolar interstitium. This helps with collagen digestion. 16 This is the best-case scenario, and animals are able to restore their pulmonary function. These cases are rarely seen in the postmortem room and normal physiology is restored in less than a week. Severe cases may die of ARDS.
If the damage to the pulmonary parenchyma is transient and there is no interference with the host ability to repair, the entire process of damage, degeneration, necrosis, inflammation, and repair should occur in less than 10 days. But, in some cases in which the animal survives and the source of alveolar and endothelial injury persists, alveolar exudation, inflammation, and proliferation can become ongoing processes that can lead to the organization of the exudates. 103 In addition to hyaline membranes, alveolar septa are thickened with edema fluid and scattered fibroblasts and myofibroblasts. These can sometimes form a myxoid matrix, together with a sparse inflammatory cell infiltrate composed of lymphocytes, plasma cells, and macrophages. In some cases, fibrin already present in alveolar spaces in the form of hyaline membranes can be covered with hyperplastic and occasional atypical type II pneumocytes. This incorporates the fibrin into the alveolar septa, which increases the thickness of this structure. This ongoing process will lead into the final stage of DAD.
Fibrosing Stage
If the stimulus for injury persists and the integrity of the basement membranes is lost, the lung lesion may evolve into the fibrosing (chronic) stage, with alveolar collapse, fusion of basement membrane, and activation of fibroblasts. 87 At this final phase, the lung is irreversibly scarred and gas exchange is no longer efficient. The main mediator for this stage is TGF-β, which drives the activation and phenotypic differentiation of fibroblasts and feeds ongoing cycles of further TGF-β production.70,72 The histologic hallmark of this stage is expansion of the alveolar interstitium, with disruption of the lung architecture and basement membranes by a combination of organized granulation tissue, with or without intra-alveolar fibrosis, and the presence of fibroblasts and myofibroblasts that are apoptosis resistant, as well as lymphocytes, plasma cells, and macrophages. 113 The vasculature of the lung parenchyma changes, with both blood vessels loss and angiogenesis driven by the local production of vascular endothelial growth factor and platelet-derived growth factor (VEGF and PDGF). In alveoli, hyaline membranes are not prominent. Frequently, there is hyperplasia and persistence of type II pneumocytes, with segments of squamous metaplasia (Fig. 3). In some areas, fibroblasts and myofibroblasts in the alveolar wall migrate through breaks in the alveolar basement membrane into the fibrinous alveolar exudate, forming intra-alveolar granulation tissue by attaching to the luminal side of the epithelial basement membrane. 56 Fibronectin, produced by alveolar macrophages, is partially responsible for the recruitment and migration of myofibroblasts in intra-alveolar spaces, and its migration to the extracellular matrix.14,27,139 Type II pneumocytes also migrate over the surface of granulation tissue and transform intra-alveolar exudate into interstitial tissue, adding to the already thickened alveolar septa. 56 This mechanism is known as fibrosis by accretion (Fig. 4). It is particularly important in experimentally induced paraquat intoxication, in which intra-alveolar fibrosis is more important than interstitial fibrosis, with resulting obliteration of alveoli. 55
One of the most important features of the fibrosing stage is the proliferation of fibroblasts and myofibroblasts in the alveolar interstitium. Fibroblasts comprise 30% to 40% of the cells in an adult normal lung and are responsible for secreting the extracellular matrix for the alveolus, which is very minimal. If there is damage and they are activated to myofibroblasts, they can proliferate and act as major contributors to fibrotic lung disease through matrix production and α-smooth-muscle-actin-mediated contractile phenotype. This is highly regulated by TGF-β. Myofibroblasts are not typically found in healthy connective tissue. 72 In conjunction with preceding tissue inflammation, changes in extracellular matrix biochemistry and/or mechanics that exhaust the homeostatic capacity of fibroblasts cause them to become activated and to phenotypically transition into myofibroblasts. 72 This is an oversimplified approach to the classification and function of the different types of fibroblasts in the pulmonary interstitium. 164 The description of each of the fibroblast subtypes, their molecular mechanisms, and interactions is beyond the scope of this review and can be found elsewhere.
There are several theories and studies about the origin of myofibroblasts. 132 One hypothesis is that lung myofibroblasts originate from a circulating hematopoietic cell, 67 but results from in vivo models are inconclusive and currently do not support this theory.31,93 The general consensus is that myofibroblasts originate from a source within the lung. 31 An aberrant epithelial mesenchymal transition has also been proposed as origin for myofibroblasts, but it has been established with in vivo models that this phenomenon reveals no significant contribution to the mesenchymal pool. 141 New information reveals that type II pneumocytes promote a profibrotic microenvironment through paracrine signaling, with activation of local fibroblasts to become myofibroblasts. 71 There are also data suggesting that endothelial cells can undergo endothelial-mesenchymal transition in pathological lung conditions, but there is not enough evidence to fully support this theory. 31 Another theory is that lung myofibroblasts may originate from perivascular mesenchymal stem cells (pericytes). Results are controversial, as previous in vivo lung studies suggested that that pericytes may proliferate with lung damage, but they do not contribute to the myofibroblast population. 141 Nevertheless, it was later concluded that the lung contains a large population of Foxd1 progenitor-derived pericytes, which is an important population of myofibroblast precursor in the lung. 75 Currently there is no in vivo evidence supporting a foreign origin or a metaplastic change in the pulmonary myofibroblasts, so the use of the term “metaplasia” (instead of hyperplasia) as a morphologic descriptor should be avoided, as such a phenomenon has not been irrefutably proven.
Acute exacerbations of diseases that have already achieved a chronic stage have been documented. These are frequently associated with invasive lung procedures, or other factors aside from congestive heart failure or fluid overload. The lesion of this phenomenon is characterized by areas of acute DAD admixed with areas of fibrosis. 2
Not Always a Diffuse Lesion
One important aspect to remember is that DAD can be patchy in the pulmonary parenchyma. The distribution depends on the cause and its distribution. Uremia, acid aspiration, radiation, and viral infections can induce DAD that is unevenly distributed within the lung. 160 For example, in calves affected with bovine respiratory syncytial virus (BRSV, bovine orthopneumovirus), lesions are frequently denser in the cranioventral aspect of lung, which is explained by the aerogenous route of infection of the virus and the absence of collateral ventilation. 23 Uneven distribution can also be observed at the margins of lesions with extensive damage to the pulmonary parenchyma such as tumors or focal areas of chronic inflammation. 160 In humans, “regional alveolar damage” has been described, which is defined as one or more macroscopically visible area of consolidation occupying less than 50% of both lungs. The underlying cause appeared to be the same to those cases with classic DAD. 187
Miscellaneous Features of ILD: Bronchointerstial Pneumonia and Acute IP in Humans
Bronchointerstitial pneumonia is a microscopic pattern, not a gross pattern. 103 Cases presenting this pattern are characterized by both direct injury to the airway epithelium and interstitial pulmonary lesions, involving the distal bronchioles and alveolar ducts with primary epithelial necrotizing injury and secondary nonsuppurative inflammation and repair. The term is also used to describe diseases in which mononuclear cells encircle airways and infiltrate alveolar septa. Bronchointerstitial pneumonia is characteristic of Mycoplasma hyopneumoniae infections in pigs and viral infections targeting cells in distal airways and alveoli, such as BRSV, canine distemper, and influenza.26,103
Bronchointerstitial pneumonia is not an all-purpose descriptor that can be used as a “wildcard” for situations with ambiguous histologic lesions that include pulmonary inflammation. If an inflammatory lesion of the lung shares features with more than one microscopic pattern, it is correct to call it “pneumonia” and to mention the features. The exercise of writing down the morphologic diagnosis is often helpful in determining the main morphologic pattern.
In human pulmonary pathology, acute interstitial pneumonia (AIP) is a clinical syndrome that manifests similar to ALI/ARDS. Confusion arises when an extrapolation of human definitions of these syndromes is attempted in veterinary cases. In human medicine, “AIP,” also known as “Hamman-Rich syndrome,” is a rare and severe form of idiopathic ILD. It is characterized by acute onset of respiratory failure, bilateral lung infiltrates on radiographs, DAD on lung histopathology, and absence of an identifiable cause or predisposing conditions. 110 Histologically, the lungs of patients with AIP display an acute or organizing form of DAD that is indistinguishable from histological patterns found in ARDS. Despite these similarities, AIP develops without any known predisposing factor, which is the main difference with ARDS.21,161 In addition to its use as a morphologic diagnosis, AIP is used to define a specific clinical syndrome. Veterinary pathologists should be cautious in applying the terminology of human lung pathology to animal diseases, because of differences from the classifications of human lung disease and the fact that “AIP” can be a clinical syndrome or a morphologic pattern of lung lesions.
Causes of DAD
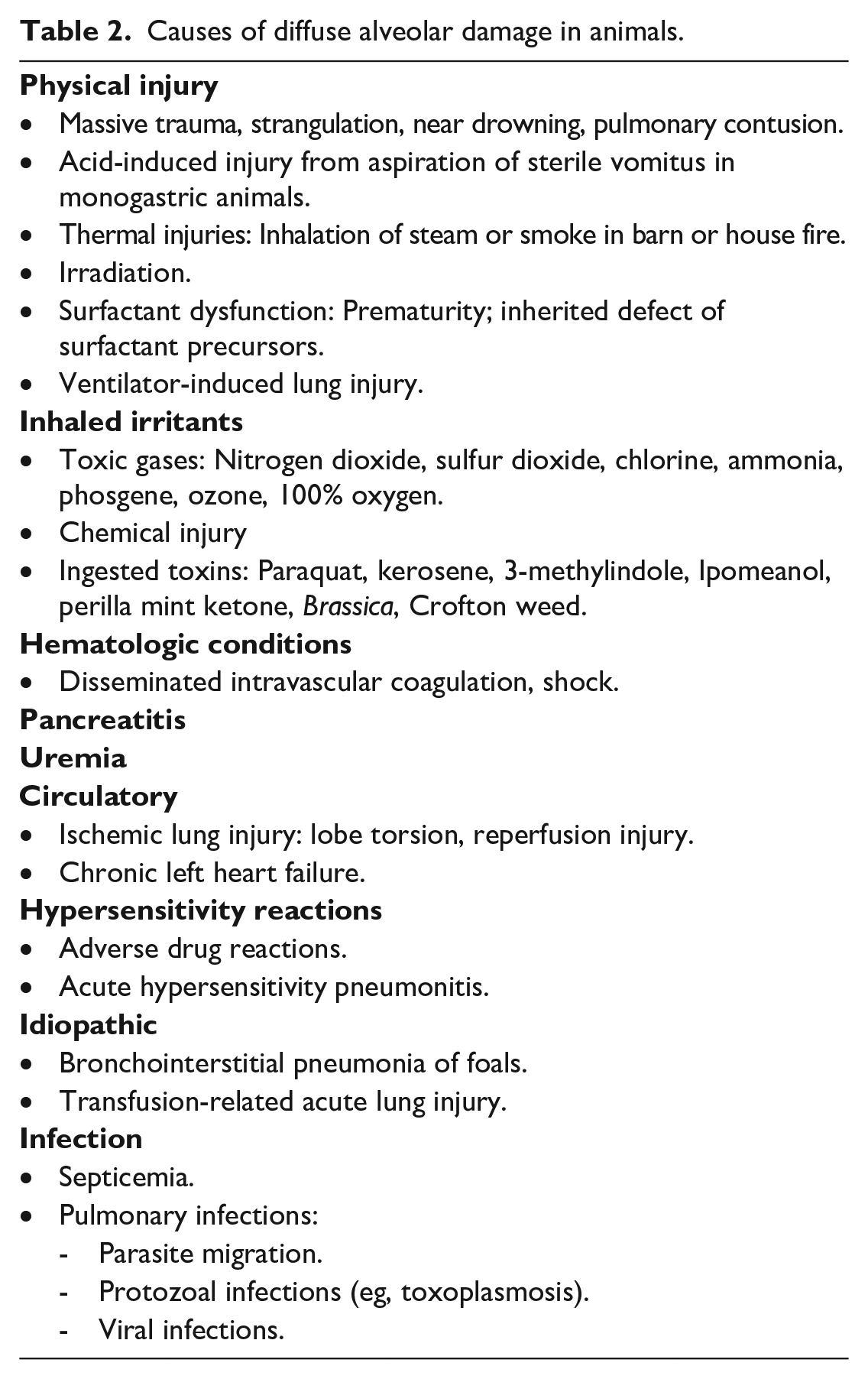
The causes for DAD are multiple, can be classified as infectious and noninfectious, and are summarized in Table 2. In human medicine, the cause of DAD can usually be inferred from the clinical history and the laboratory results. Communication between the clinician and pathologist is essential to the establish the probable cause of DAD. 25 This fact acquires even more importance when the histologic examination of the lung does not provide much etiologic information.
Causes of diffuse alveolar damage in animals.
One of the most important processes of DAD is the generation of inflammatory mediators for the initiation and progression of this condition, known as the cytokine storm.
Cytokine Storm
The term cytokine storm or cytokine release syndrome refers to an activation of the immune system beyond the degree sufficient to control an event, such as viral or bacterial infection, non-self-tissue engraftment, or exposure to certain therapeutic drugs.83,151,159 Usually, upon infection, the immune system will detonate a proportional response against the agent allowing the host to survive the episode. However, cytokines have the potential of inducing injury if this release is not controlled or if it lasts for a prolonged period of time. 107 During cytokine release syndrome, the immune response generates collateral damage that is greater than the benefit from the immune response. This is often characterized by elevated cytokines levels, acute systemic inflammation, and organ dysfunction. 48 Pro-inflammatory cytokines including IL-6, IFN-γ, and TNF-α have been implicated by numerous authors as mediators of cytokine release syndrome. The extent of their elevation depends on the nature of the insult.83,152,159,172 Action of these mediators can lead to pulmonary edema, vascular leakage, and ARDS, resulting in the need for intensive care.151,171,172
As described previously, different viruses, bacteria, and stressors can all inflict injury at different sites of the lung and can detonate the cytokine cascade by stimulating pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs), as well as RIG I RNA helicase signaling or Nod-like receptors. 159 Initially, IL-1β and TNF appear at the early phase of infection and stimulate the production of IL-6. In a well-regulated response, negative regulators such as IL-10 produced by macrophages and T-regulatory cells would temper the inflammatory response.48,82,159 However, in cytokine release syndrome, multiple factors fail to return the system to homeostasis, including the nature of the pathogen/insult, hyperresponsive receptors for pathogen-associated molecular patterns, the abundance of pro-inflammatory cytokines and chemokines, and inadequacy of the anti-inflammatory response.48,82,159
TNF is a strong pro-inflammatory cytokine that modulates antimicrobial defense, stimulates IL-6 production, and activates multiple pro-inflammatory genes. Macrophages and dendritic cells contain an intracellular sensor called the inflammasome that recognizes pathogens and stressors and stimulates the production of IL-1 and IL-18. These two then stimulate the production of IL-6 and synergize with other pro-inflammatory molecules. IL-6 can activate different pathways even in cells that lack the surface receptor IL-6R, such as endothelial cells.48,159,171 This cascade contributes to cytokine release syndrome by systemic increase of monocyte chemoattractant protein 1 (MCP-1), IL-8, IL-6, and VEGF, and reduced E-cadherin on endothelial cells. Systemic inflammation is thus associated with vascular hyperpermeability, fibrin deposition, disseminated intravascular coagulation (DIC), depletion of coagulation factors, and pulmonary and kidney dysfunction.48,74,82,107,159 In addition, excessive activation of complement can similarly result in endothelial damage, contributing to dysregulated coagulation and loss of vascular tone. Vascular endothelium from lung and kidney is generally more susceptible to complement-mediated injury. 107
DAD Noninfectious Causes
Physical Injury: Ventilator-Induced Lung Injury
Ventilator-induced lung injury is indistinguishable morphologically, physiologically, and radiologically from other causes of DAD. 170 A simplistic explanation is that lung damage by mechanical ventilation occurs due to the cyclic stretch of the airway when there is an infection in the lung, but it can also occur without infection, 127 leading to overdistention of alveoli and injury to the septa.
Several authors have described ventilator-induced lung injury as happening either at the initial anisotropic alveolar inflation or during lung collapse at the end of the expiratory phase.22,127 The trauma can be further classified as volutrauma, in which overdistention of alveoli causes stretch-induced damage to cells; barotrauma, which is direct damage induced by the high pressure in the lung during mechanical ventilation; atelectotrauma, which is trauma from repetitive opening and closing/collapse of the alveoli; and biotrauma, which is mediated by inflammatory mediators in the lung.22,170 The trauma induced during the initial anisotropic phase promotes a local inflammatory response. This can lead to endothelial disruption, which is a major feature DAD caused by ventilator-induced lung injury; this endothelial damage precedes pulmonary edema and leads to neutrophil infiltration.9,127 Other features are increased surface tension due to reduced surfactant function and direct injury to pneumocytes by tensile and interfacial stress. 22 Tissue deformation activates nuclear factor-kappa B signaling, leading to the production of IL-6, IL-8, IL-1β, and TNF-α. 170 The main consequences of the edematous fluid in the alveoli include its interference with pulmonary surfactant, elevating surface tension, and reduced clearance of fluid. In addition, the overproduction of these pro-inflammatory cytokines can lead to massive neutrophil infiltration. 133 The lung regions that are left poorly ventilated then become inactive and no longer take part in gas exchange. 9
Thermal and Chemical Injury
Thermal injury occurs in livestock during barn fires, but can also occur to any other species. 103 It is not uncommon for patients with severe burn injuries to have associated smoke inhalation injury. This type of injury is first inflicted by direct thermal damage to upper respiratory system tissues with subsequent increase of inflammatory mediators and vascular permeability, leading to edema and migration of neutrophils exacerbating the chemotactic response. Should the mucociliary system become affected, there might occur distal migration of mucus, fibrin, cellular debris, and bacteria that can lead to infection. 168 Thermal injury leads to ALI by activation of the complement system (C5), circulating neutrophils, and toxic oxygen-derived radicals, all leading to endothelial damage. 4
Similarly, gastric acid–mediated injury initially induces direct damage to the mucociliary system and alveolar epithelium. The extent of lesions induced by gastric acid depends on the volume of the aspirate and on the pH. Gastric acid prevents the growth of bacteria, therefore the content of the stomach is sterile. 157 Aspiration of gastric acid triggers a cascade of inflammatory responses with the recruitment of neutrophils and release of various inflammatory mediators, such as TNF-α and CXC chemokines. 108 Morphologically, it is characterized by DAD, with hyaline membranes, alveolar hemorrhages, and neutrophils. 174 the aspiration of gastric contents reduces the host defenses against infections, predisposing to secondary bacterial infections. 145
Another frequent source of chemical injury is ammonia inhalation. Ammonia is used as fertilizer in agriculture and is produced as a natural biological process in humans and animals. Urea is water-soluble, and when in contact with water-rich mucous membranes it forms ammonium hydroxide, which is a highly caustic compound. 54 The upper respiratory tract is commonly affected with deciliation, necrosis, and ulceration of nasal passages and tracheal mucosa.54,191 When the exposure is considerable, it can induce lung injury characterized by cranioventral necrotizing bronchiolitis, pneumonia, and prominent type II pneumocyte hyperplasia with occasional multinucleated giant cells. 54 In humans accidentally exposed to ammonia, corrosive burns on the oropharynx and DAD are identified, together with high level of ammonia in blood. 166
“Fog fever” occurs in cattle moved from dry pastures to pastures that include alfalfa, kale, turnip, and grasses rich in
Hematologic Conditions: Systemic Inflammatory Response Syndrome and Disseminated Intravascular Coagulation
There are multiple clinical conditions, including infections, disseminated neoplasia, or diseases with major tissue destruction (eg, major trauma, pancreatitis, systemic inflammation), that can activate the coagulation system.57,96 Some of these events may lead to exuberant activation of hemostasis that may present as DIC, which is characterized by the synchronized occurrence of extensive microclot formation.58,96 Briefly, there is initiation of coagulation by tissue factor exposure to circulating blood, increased interaction of platelet and vessel wall, impaired regulation of coagulation, and defective fibrinolysis. 96 Endothelial cells, which are considered as innate immune cells and can secrete inflammatory cytokines, can be damaged, causing defective vasoregulation, inflammation, and induction of oxidative stress, with consequent multi-organ failure. 91 There is significant bidirectional cross-communication between inflammatory activation and hemostatic activity. 97 In cases of impairment of pulmonary microcirculation, hypoxia can lead to endothelial damage that can be accompanied by neutrophils within the alveoli and septa. 128 At the same time, vascular dysfunction can promote an inflammatory host response, with excessive activation of immune cells and cytokine storm, which promotes a hypercoagulable state and thrombosis. 33 Two hemostatic mechanisms have recently increased the understanding of ARDS and DAD from its molecular pathogenesis in the development of an endotheliopathy: the “two-activation theory of the endothelium,” characterized by endothelial activation of inflammatory and microthrombotic pathways, and “the two-path unifying theory” of hemostasis, in which hemostasis initiates thrombogenesis and promotes microthrombogenesis leading to vascular microthrombotic disease.28 –30 Both theories include contributions of endothelial cells to initial hemostasis and triggering the molecular mechanism of thrombogenesis.
Severe cases of pancreatitis, commonly known as acute necrotizing pancreatitis or acute pancreatic necrosis, can lead to systemic inflammatory response syndrome, and a few cases can result in multiple organ dysfunction syndrome. 169 However, among all of the systemic complications arising during acute necrotizing pancreatitis, respiratory complications leading to ARDS with DAD are the most severe.61,169 In humans, this syndrome is called pancreatitis-associated lung injury and occurs in up to 75% of cases of severe acute pancreatitis, leading to death in approximately 40% of the cases.61,190 In contrast, in dogs, clinical respiratory complications occur in only 7% to 23% of acute pancreatitis cases. 169
Pancreatic enzymes released during pancreatitis contribute to the pro-inflammatory cytokines release with activation and migration of leukocytes in the pancreas.61,149,190 Activated pancreatic macrophages induce high levels of IL-8 and TNF-α. 149 After the initial damage to the pancreas, there is release of trypsin that causes direct damage to the lung vasculature; activates mediators that include substance P, elastase, and phospholipase A2; and leads to further lung injury by increasing endothelial permeability, inducing pulmonary endothelial injury. DAD is a consequence of systemic inflammatory response leading to increased permeability of the endothelial and epithelial barriers, with leakage of protein-rich exudate into the alveolar space.150,189
Dogs can develop respiratory complications of acute pancreatitis. One report described lung inflammation with increased levels of IL-6, TLR 4, and iNOS as well as recruitment of PIMs in dogs with necrotizing pancreatitis. 169 In addition, necrosis and exfoliation of epithelial cells, edema, abundant neutrophils, and macrophages and very characteristic thick hyaline membranes lining the alveoli have been described in dogs with pancreatitis, with the diagnosis of DAD. 102
Uremia
Uremic pneumonopathy occurs in dogs with chronic renal failure and often leads to mineralization of the smooth muscle and connective tissue of capillaries and alveolar septa in the pulmonary interstitium. 94 Pulmonary mineralization has been associated with azotemia in 60% to 80% of the human autopsy cases of chronic renal failure. 39 Chronic kidney disease can cause a variety of lung alteration such as interstitial edema, pleuritis, pneumonia, vascular damage, and alveolar damage. These changes are mostly due to increased permeability, channel dysregulation, increased cytokines and chemokines, and inflammatory response.39,103 Briefly, during chronic renal failure there is retention of phosphate and hyperphosphatemia that can form complexes with calcium and mineralize alveolar septa and other tissues. 94 In addition, uremic toxins can induce direct damage to pneumocytes by increasing ROS and altering the function of epithelial sodium channels, Na2+/K+-ATPase downregulation, and aquaporin dysfunction, which oversee maintenance of sodium absorption, water transport, and fluid clearance in the alveoli, all relevant during DAD.99,135,185
Circulatory Causes of DAD
There are not many events that can lead to ischemic lung injury, due to the dual circulation of the lung.26,134 Nevertheless, there are reports in the veterinary and human literature associating the presence of DAD resulting from pulmonary thromboembolism, which in some cases may lead to pulmonary infarction.86,162 Sublethal ischemia may cause histologic lesions of DAD, whereas infarcts manifest as foci of coagulative necrosis. 26 The precise mechanism of DAD in these cases is not fully elucidated, but it has been associated with inflammatory cytokines and chemokines, neutrophils, and platelet aggregates.86,188 It is suggested that pulmonary arterial hypoperfusion is sufficient to induce a pro-inflammatory status, and that inflammatory mediators might be responsible for pulmonary thromboembolism-related DAD. 86
Another well-known cause for a similar set of findings occurs secondary to ischemia-reperfusion injury after lung transplantation in humans. It is possibly caused by an excess of ROS, endothelial cell injury, increased vascular permeability, activation of neutrophils and platelets, complement system, and inflammatory cytokines.44,50
It is well known that patients with cardiac disease or heart failure have a high incidence of pulmonary infarction following embolization.26,162 In addition to embolization, late stages of congestive left heart failure can result in pulmonary venous hypertension. This is accompanied by increased capillary hydrostatic pressure and a series of events that damage the alveolar wall, including hydrostatic mechanical injury of endothelial cells and alveolar cell detachment, impairment of the cellular pathways involved in fluid reabsorption, and resistance to gas transfer.63,64 Alveolar edema is the most impressive consequence of stress failure, together with increased numbers of lung macrophages and remodeling of pulmonary veins.26,178 Remodeling of the alveolar wall with arterial changes only occur in very late stages of left heart failure. DAD is sometimes observed in simple congestive left heart failure. An animal model has provided evidence that DAD can develop as a consequence of left heart failure in cattle, in which hyaline membranes and type II pneumocyte hyperplasia were multifocally evident in the lungs from all experimental animals. 126
Hypersensitivity Reactions
The lung is a major anaphylactic organ in cattle. Anaphylaxis or anaphylactoid reactions due to intravenous administration of different compounds (eg, inadvertent intravenous inoculation of vaccines, endotoxin) can affect the lungs in cattle, causing a rapid death after a short episode of bronchoconstriction, pulmonary hypertension, and systemic hypotension.26,47 Clinically, it is characterized by severe dyspnea with red foamy nasal discharge. 137 At necropsy, lesions include pulmonary congestion, edema, and emphysema. Histologic lesions are characterized by multifocal to coalescing severe intra-alveolar and interstitial hemorrhages.47,137 Findings in bronchi and alveoli are proteinaceous edema with moderate numbers of macrophages and infiltrates of neutrophils, plus marked interstitial edema, periarterial hemorrhages, and occasional fibrin thrombi.26,137,154
Hypersensitivity pneumonitis (or “extrinsic allergic alveolitis”) is a group of immunologically mediated lung diseases due to the repeated inhalation of finely dispersed antigens of a wide variety, capable of triggering mononuclear to granulomatous inflammation in distal bronchioles and alveoli of susceptible subjects.17,20 Hypersensitivity pneumonitis in cattle has been associated with chronic inhalation of spores of thermophilic actinomycetes (especially Saccharopolyspora rectivirgula) in moldy hay.26,180,181 Exposed animals displayed noncollapsed lung lobes, edema, emphysema, and cranioventral consolidation. In chronic cases, septal infiltration of mononuclear cells to the point of obliteration of alveolar lumen, type II pneumocyte hyperplasia, conglomerates of alveolar macrophages in the alveoli and interstitial proliferation of connective tissue were noted, similar to the changes described in cases of fog fever, anaphylaxis, or other causes of alveolar damage.180,181 The mechanism of damage is direct activation of the complement pathway by the antigen, plus a type III and IV hypersensitivity.26,181 In human pathology, findings associated with hypersensitivity pneumonia are centered on bronchioles and include lymphocyte-rich infiltrates around bronchioles (bronchiolitis) or poorly formed non-necrotizing granulomas within the peribronchiolar interstitium. Particularly, for bronchiolitis, it may include peribronchiolar fibrosis and hyperplasia of the bronchiolar epithelium (referred as “peribronchiolar metaplasia”), which are common but not specific findings. 123
Hypersensitivity pneumonitis has also been experimentally reproduced in rats exposed to pigeon droppings, in an attempt of creating a model for pigeon breeder’s disease. In these animals, there were interstitial infiltrates of lymphocytes, plasma cells, and foam cells, the latter also present in alveoli. 53
DAD—Infectious Causes
Lungs are a common a target for numerous airborne and hematogenous pathogens, and the development of DAD with a strong inflammatory response can lead to subsequent ALI/ARDS. Lesions can be induced by sepsis, parasitic infections, and viral infections.
Sepsis
Sepsis is one of the leading causes of DAD for which the innate immune system plays an important role, 92 and in veterinary medicine the leading cause of sepsis is bacterial infection. 46 Sepsis-associated DAD can be induced by direct effects of bacterial infection in the lung, with epithelial damage resulting from bacterial toxins or as a result of the inflammatory response.43,84,186 Alternatively, sepsis-induced DAD can be indirect, arising from extrapulmonary infection that leads to alveolar-capillary barrier dysfunction, by injury to the endothelial cell surface, formation of platelet-leukocytes aggregates, alteration of the glycocalyx, and release of inflammatory mediators.43,84
Once bacteria are recognized by macrophages and PRRs on the resident immune cells, the production of pro-inflammatory cytokines begins. IL-1 and TNF-α stimulate production of IL-6 and IL-8. Then, neutrophils are attracted to the site of infection and can lead not only to pathogen clearance but also to direct pulmonary damage to alveolar and bronchiolar epithelial cells. At the same time, PRRs from natural killer T cells initiate the release of cytokines and chemokines, boosting the neutrophil infiltration. The cytokine response to the microbes leads to loss of alveolar-capillary barrier integrity, neutrophil recruitment, surfactant dysfunction, and alveolar edema.43,98
Another important component of sepsis-associated DAD is the increase in plasminogen activator inhibitor type 1, inhibiting the fibrinolytic pathway and thus contributing to widespread microvascular thrombosis. This can lead to DIC in severe cases. 43
PIMs are especially important in cases of sepsis in animals. PIMs are a resident population of mature macrophages that adhere to endothelial cells in pulmonary capillaries. 114 PIMs are present in some animal species, such as horses, cattle, sheep, goats, cats, pigs, and cetaceans. Particulate matter and lipopolysaccharides in blood are recognized by PIMs in the lungs, which makes these species more susceptible to lung injury. This is considered a major factor in the sensitivity of these species to endotoxin-related lung injury.18,175 PIMs have a robust phagocytic capacity and are able to secrete oxygen radicals for microbicidal function. 184 Compared with alveolar macrophages, they are equally phagocytic and cytolytic for bacteria and produce cytokines (IL-1) and TNF-α, but are more cytolytic for virus-infected cells. 34 PIMs also have a role in the removal of erythrocytes, fibrin, cellular debris, and immune cells from the circulation. 148 There is communication between the alveolar and capillary environment across the alveolar septum, and PIMs can amplify the signals, which can result in higher levels of vascular inflammation. 90
Parasitic
Among many infectious causes of DAD, parasitic infections are frequent in veterinary medicine. Parasites such Dictyocaulus spp. and Toxoplasma spp. are able to infect the lung and thus are most commonly associated with this pattern of lung injury.
Dictyocaulus spp. affect ruminants (D. viviparus) and horses (D. arnfieldi) and has also been reported in wild animals such elk, moose, and deer (D. cervi, D. capreolus, and D. eckerti).6,52,130 Infections occur via a direct cycle where the adult nematodes deposit eggs in the bronchi and bronchioles, from which hatching L1 larvae ascend, and are swallowed and excreted with the feces into the environment. There they mature to L3 for D. viviparus and L2 for D. arnfieldi and are once again ingested, turning into L4 at the mesenteric lymph nodes and migrating into the lung as L4. 59 Helminths’ chitin, shed during molting, along with other glycans and lipids molecules from the parasite, acts as pathogen-associated molecular pattern recognized by the host immune system.105,140,176 In addition, helminths release extracellular vesicles that contain proteases, glycolytic enzymes, and lectins that contribute to the direct tissue damage. 176 During the acute phase of larvae migration, epithelial cells secrete alarmins leading to the production of type II–associated cytokines (IL-4, IL-5, IL-9, IL-10, and IL-13); chemokines such as CX CL1, CXCL2, CXCL8, and eotaxin, which signal innate immune cells such as basophils, eosinophils, neutrophils, and macrophages, typically resulting in initial goblet cell hyperplasia, increased mucus secretion, and eosinophilia, with high levels of IgG and IgE and can easily lead to uncontrolled production of these cytokines and decreased anti-inflammatory mechanisms.60,140,176 In contrast, the persistent infection can induce a T-helper 1 response resulting in production of IL-2 and IFN-γ and expansion of IL-10–secreting T-reg cells. 60 DAD with the presence of larvae has been identified, together with mononuclear cell infiltration of the alveolar septa. In chronic cases, severe pulmonary fibrosis and lymphoid hyperplasia have been described. 52
Another parasitic infection that can lead to DAD is Toxoplasma spp., an intracellular apicomplexan protozoan widely distributed around the world. Toxoplasma infections induce IL-12 production by the innate immune system and consequent release of IFN-γ from NK and T cells and neutrophils. 42 The lytic cycle, where the parasite undergoes asexual growth and multiplication, induces direct cell damage by rupture and release of the tachyzoites. 78 Rapid proliferation of tachyzoites can lead to a strong activation of the immune system mediated by neutrophils, eosinophils, and mononuclear cells, causing DAD, especially in immunocompromised hosts or when acquired by congenital infection.37,40,46,125 Histologically, DAD is characterized by macrophages and fibrinous exudate filling alveoli, plus multifocal necrosis of the alveolar epithelial cells, alveolar epithelium, and blood vessels. These lesions are followed by type II pneumocyte hyperplasia and hypertrophy, which can confer an adenomatous appearance. Tachyzoites can be found in alveolar macrophages, bronchiolar epithelial cells, and within walls of blood vessels. 165
Viral
Viral pneumonias in animals are frequently associated with interstitial patterns of inflammation of the lung, particularly with DAD. 26 Among many animal species, DAD can be caused by virus infections that include influenza virus, parainfluenza virus, herpesvirus, adenovirus, feline calicivirus, respiratory syncytial virus, and coronavirus, among others.32,35,62,76,142 Viruses can cause DAD through direct and indirect mechanisms. Direct damage is characterized by direct cytolytic damage to bronchiolar epithelial cell, types I and II pneumocytes, and endothelial cells. Indirect damage is frequently reached by cytokine-induced permeability of the air-blood barrier and excessive recruitment and activation of inflammatory cells, particularly macrophages and neutrophils.36,124 The persistence of a viral infection and the antiviral immune response frequently result in widespread pulmonary damage and secondary complications, such as bacterial coinfections and systemic inflammation, due to dysregulated immune response. 36
Severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV), respectively, emerged in humans in 2002 and 2012 and caused severe pneumonia that rapidly progressed in some cases to ARDS.32,62 Similarly, severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) emerged in 2019 in Wuhan. 118 In this case, the clinical signs vary from minimal to severe, with ARDS reported in up to 15.6% of the patients with respiratory disease. 118 Upon binding, coronaviruses downregulate ACE2 expression in the infected host cells. This leads to an increase in angiotensin II and contributes to lung injury.32,68,121 It has also been proposed that by loss of inhibition in the phosphorylation of JNK, there is activation of caspase 9, suggesting that apoptosis occurs by the mitochondrial pathway. 163 Infected cells express cytokines including IL-6, IL-8, TGF-α and TGF-β, and monocyte chemotactic protein 1. 68 Other factors such as IL-6, IL-7, and TNFs play an important role in SARS-CoV2 infection and can contribute to the cytokine storm. 118
SARS-CoV2 infection commonly has other features that contribute to severe disease manifestation, such as DAD, extravascular fibrin deposition, neutrophil trapping, microvascular thrombosis, and large vessel pulmonary emboli. 76 In human cases, DAD triggered by SARS-CoV-2 infection is morphologically indistinguishable from other causes of DAD. 88
DAD-like lesions are reported in some SARS-CoV-2 studies in laboratory animals,95,122 but lung lesions in these experimental studies are often not typical of human or domestic animals’ DAD. Other papers in this special issue will discuss lung lesions in those experimental animals. SARS-CoV2–induced DAD-like lesions in Roborovski hamsters and mink most closely resemble those in humans (in this issue in Gruber et al and Ritter et al).
Conclusions
IP due to DAD is a common reaction to lung injury, especially when this injury is directed to the alveolar epithelium and/or endothelial cells. There is a well-known progression of stages, with histologic features that are characteristic for every level of progression. Complications arise in the interpretation of the findings and diagnosis, mostly when there are numerous causes with similar lesions. A further complication is the common occurrence of secondary bacterial infection, which add more cells to the lesion, and hence, more confusion about the pathogenesis at the time of interpretation. Regardless of the cause, the progression of DAD is mediated by inflammatory mediators that trigger an inflammatory reaction, which is exacerbated by secondary damage due to the arrival of inflammatory cells. From a mechanistic perspective, the development of DAD is very similar in humans and animals. Regrettably, veterinary medicine is far from a comprehensive understanding of the different disease syndromes that cause DAD, mostly because of the lack of communication between the clinicians, radiologists, and pathologists. With this in mind, it becomes relevant that veterinary pathologists share a similar terminology, so the communication channels remain open to the advancement of the diagnosis of respiratory diseases in animals.
Footnotes
Acknowledgements
We thank Dr Phillip Sponenberg and Dr Thomas Cecere for the useful comments about the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.