
Abstract
Balancing cell survival and cell death is fundamental to development and homeostasis. Cell death is regulated by multiple interconnected signaling pathways and molecular mechanisms. Regulated cell death (RCD) is implicated in fundamental processes such as organogenesis and tissue remodeling, removal of unnecessary structures or cells, and regulation of cell numbers. RCD can also be triggered by exogenous perturbations of the intracellular or extracellular microenvironment when the adaptive processes that respond to stress fail. During the past few years, many novel forms of non-apoptotic RCD have been identified, and the characterization of RCD mechanisms at a molecular level has deepened our understanding of diseases encountered in human and veterinary medicine. Given the complexity of these processes, it has become clear that the identification of RCD cannot be based simply on morphologic characteristics and that descriptive and diagnostic terms presently used by pathologists—such as individual cell apoptosis or necrosis—appear inadequate and possibly misleading. In this review, the current understanding of the molecular machinery of each type of non-apoptotic RCD mechanisms is outlined. Due to the continuous discovery of new mechanisms or nuances of previously described processes, the limitations of the terms apoptosis and necrosis to indicate microscopic findings are also reported. In addition, the need for a standard panel of biomarkers and functional tests to adequately characterize the underlying RCD and its role as a mechanism of disease is considered.
Keywords
It is extraordinary how quickly the definition of cell death has changed before our eyes. To give a sense of the pace of the current research and discoveries on cell death, a PubMed search of the term regulated cell death returned a total of 321,120 results, 56% of which were concentrated between 2010 and 2020. With all certainty, this means that by the time this review is published, some of the concepts it contains will be outdated.
Undoubtedly, the most recent breakthroughs in the study of cell death have sparked an unprecedented interest for this field of research. 124,283 At the same time, all this traction and excitement have generated confusion around emerging definitions, classifications, and terminology to describe the overwhelming surge of novel “-ptosis” modalities. In a very interesting perspective article, Liu and colleagues criticize the “chaos” caused by the expanding cell death vocabulary. The authors identified at least 34 diverse designations of cell death modalities, most of which have been coined only very recently. 193 In this rapidly evolving scenario, it is important to keep in mind that current research efforts are just scratching the surface of the cell death conundrum.
While the biological significance for some of the latest discoveries in the cell death arena have been established (eg, necroptosis, pyroptosis, and neutrophil extracellular trap [NET]-otic cell death), there are many new paradigms that are still in search of a formal proof of concept outside the Petri dish. This is especially true for those cell death types whose demonstration relies entirely on factitious in vitro experiments (eg, ferroptosis and mitochondrial permeability transition (MPT)-driven necrosis). In addition, as we learn more about the mechanisms of cell death, it is becoming clear that many of the newly defined “-ptosis” types are actually profoundly connected, featuring overlapping molecular characteristics and hierarchical associations on the same pathways. 105,121,224,328 Therefore, it is reasonable to expect that closely interrelated processes simply represent nuances of the same mechanism and will be eventually grouped under a common cell demise paradigm.
As the cell death field continues to grow and novel signaling pathways are unveiled, the Nomenclature Committee on Cell Death (NCCD) is proactively taking steps to mitigate some of the existing discrepancies, consolidate the emerging hypothesis, and provide an updated classification of cell death modalities. Emphasis is placed on the mechanisms involved in the initiation, execution, and propagation of cell death as well as on the pathophysiological relevance of each of the main types of cell demise (ie, cell death mechanisms whose level of characterization is sufficient to grant their classification as defined entities). 105
Within organisms, cells that are exposed to extreme physicochemical or mechanical stressors may undergo an immediate and uncontrollable structural breakdown, leading to a phenomenon referred to as “accidental cell death” (ACD). Conversely, regulated cell death (RCD) encompasses a number of processes involving specific signaling cascades and molecularly defined effector mechanisms leading to a cellular demise that can be modulated pharmacologically or genetically. 99,100,283 Historically, the first observation of cell death occurred in the 19th century, although the phenomenon was generally considered passive and unregulated. Research efforts on cell death did not start until the mid-20th century, with the first description of apoptosis occurring in 1972. 283 Since then, due to the active discovery of novel signaling pathways that contribute to RCD modalities, the NCCD has formulated guidelines and consensus criteria for the definition and interpretation of cell death. 105,176 While RCD is implicated in the pathogenesis of specific conditions in humans and animals, it has become clear that RCD also occurs under physiological conditions and plays a fundamental role in human and animal development, organogenesis, tissue remodeling, and tissue homeostasis. 94,99,100,283
In addition to apoptosis and necrosis, increasing interest has recently centered on other emerging RCD modalities including ferroptosis, pyroptosis, necroptosis, parthanatos, entotic cell death, NETotic cell death, anoikis, MPT-driven necrosis, lysosome-dependent cell death, autophagy-dependent cell death, and immunogenic cell death. 105 Here, we review the current knowledge pertaining to the molecular aspects and pathways of the RCD modalities that are officially recognized by the NCCD, 105 with a focus on available biomarkers, tools, and other functional tests to evaluate several aspects of RCD. Implications and remarks that are specifically relevant to veterinary pathology are also highlighted. While little is known about the role of specific RCD mechanisms in veterinary conditions, some examples of known associations will be provided. A list of abbreviations is provided in Supplemental Table S1.
The Current Histopathologic Criteria Fall Short on RCD
Early classifications of cell death modalities were focused on the recognition of microscopically identifiable changes of individual dying cells. 100,283 This has led to the distinction of 2 main cell death morphologies, namely, necrosis and apoptosis (as well as mixed or biphasic entities such as oncotic necrosis, apoptotic necrosis, and apoptonecrosis), 201 which have been incorporated into the standardized pathology terminology and largely used as descriptive and diagnostic terms without a complete understanding of the underlying molecular mechanisms. 80,178,250,251 Considering that cell death may occur in many different ways, it is noteworthy to mention that the presence of specific morphological features is not sufficient to establish the occurrence of a given process of cellular demise. A growing body of evidence has highlighted the unsuitability of this approach emphasizing that (1) necrosis is a rather nebulous and nonspecific term that can be applied to diverse types of ACD or RCD and (2) apoptosis is just one of the many manifestations of RCD. In addition, depending on the specific biological and pathological context, diverse cell death modalities may display very similar (if not identical) microscopic and biochemical features. 105,343 Therefore, although distinctive guidelines have been made available to aid in the histologic recognition and morphologic classification of apoptosis and necrosis, 81 use of a merely morphological endpoint based on the traditional apoptosis/necrosis dichotomy is largely insufficient to depict the complexity of the current cell death scenario. 87 While it is recognized that differentiation of the precise pathway of cell death may not be fundamental in pathology settings, it is important to remember that many other RCD modalities have been characterized over the years, and the field is continuously evolving. Thus, the use of terms such as apoptosis, necrosis, and single-cell apoptosis/necrosis may be misleading. With this in mind, pathologists and pertinent working groups of international initiatives, such as the INHAND (International Harmonization of Nomenclature and Diagnostic Criteria), should consider revising the existing recommendations acknowledging the fact that any description of hematoxylin and eosin (HE)-stained tissue sections that goes beyond “cell death” is, more often than not, a potential biological misinterpretation of a morphological readout. Nonetheless, pathologists’ expertise remains essential to guide the discovery of reliable biomarkers to identify different cell death modalities and unveil their biological significance in the intact tissue context as well as the correlation with ongoing pathological processes such as inflammation, circulatory disorders, degenerative conditions, and so on.
Apoptosis With Emphasis on Anoikis
Apoptosis, the best studied form of RCD, is a process that requires the activation of caspase proteases. Two major subtypes of apoptosis exist, namely intrinsic and extrinsic apoptosis (Figs. 1, 8). The intrinsic apoptosis pathway, which is physiologically dominant, is triggered by irreversible and widespread mitochondrial outer membrane permeabilization (MOMP) with release of the mitochondrial proteins such as cytochrome C (CYCS) and direct inhibitor of apoptosis-binding protein with low isoelectric point (DIABLO; also known as second mitochondria-derived activator of caspases [SMAC]). This is regulated mainly by members of the B-cell lymphoma (BCL) 2 family of proteins including pro-apoptotic proteins (eg, BCL2-associated X, apoptosis regulator [BAX], BCL2 antagonist/killer 1 [BAK1]), and anti-apoptotic proteins (eg, BCL2 apoptosis regulator [BCL2]; BCL2-like 1 [BCL2L1, also known as BCL-extra large (BCL-XL); and MCL1 apoptosis regulator, BCL2 family member [MCL1]). 105,271,283 In addition, the BCL2 binding component 3 (BBC3, also known as PUMA [p53-upregulated modulator of apoptosis]), BCL2-like 11 (BCL2L11, also known as BIM [BCL-2-interacting mediator of cell death]), phorbol-12-myristate-13-acetate induced protein 1 (PMAIP1, also known as Noxa), and BH3-interacting domain death agonist (BID) physically interact with the mitochondrial pool of BAX and/or BAK to promote conformational changes. The release of cytochrome C is eventually responsible for the activation of caspase 9 via the oligomerization of the apoptotic protease activating factor-1 (Apaf-1). The extrinsic apoptotic pathway is mediated by membrane receptors, especially by death receptors (eg, tumor necrosis factor [TNF] receptor superfamily member 6 [TNFRSF6], also known as FS-7-associated surface antigen [FAS] or cluster of differentiation [CD] 95, and TNF receptor superfamily member 1A [TNFRSF1A, also known as TNFR1]), and is propagated by caspase-8 and caspase-10. 105 Ultimately, caspases -3, -6, and -7 are considered the common effector caspases (ie, executioner caspases) for both pathways. Caspase activation can be blocked by X-linked inhibitor of apoptosis (XIAP). While excellent reviews on apoptosis are available, 80,271,285 less known subtypes of apoptosis, such as anoikis, also exist.
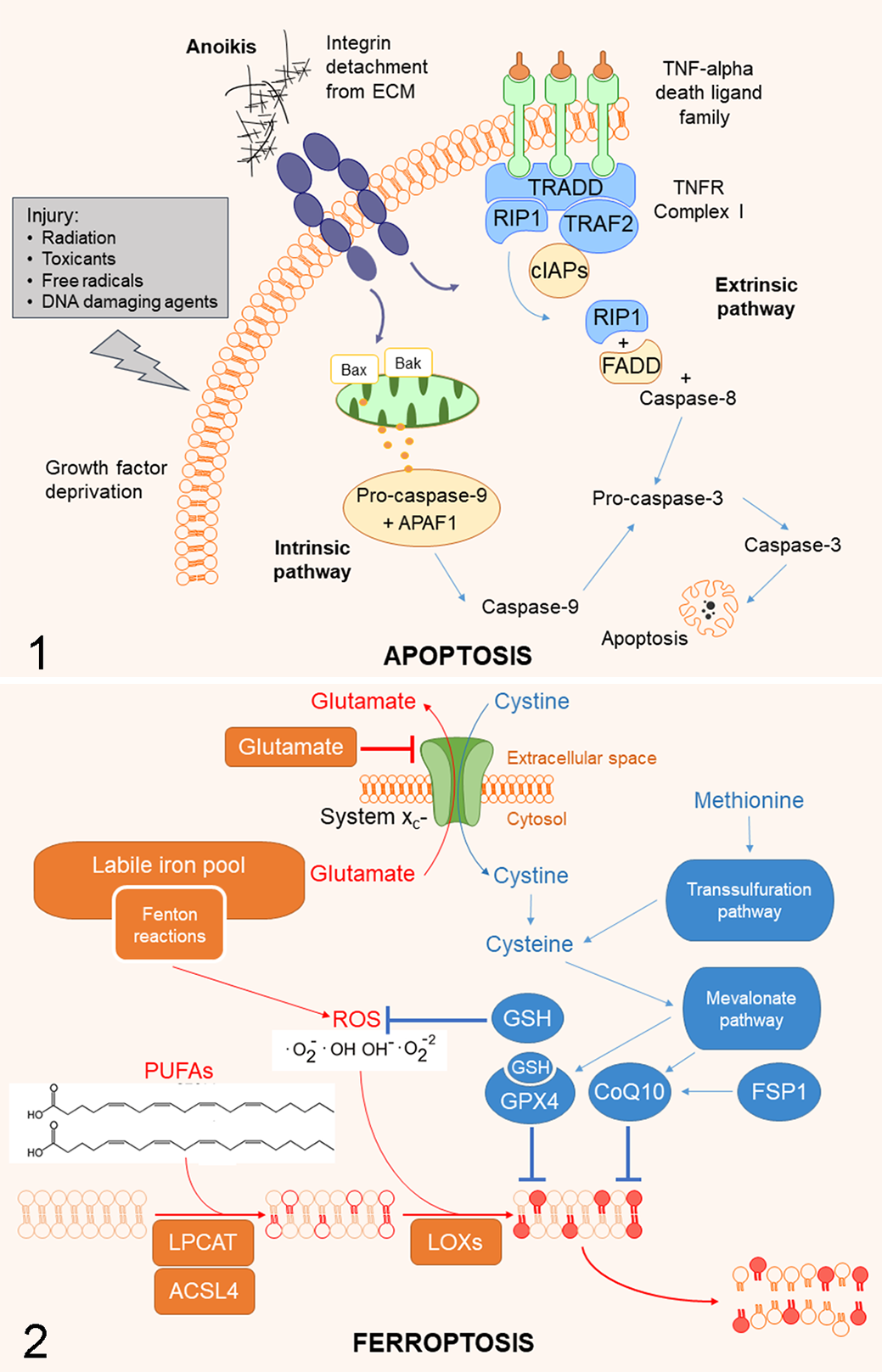
Apoptosis. In the intrinsic apoptosis pathway, mitochondrial outer membrane permeabilization with release of mitochondrial proteins is triggered by stimuli such as growth factor deprivation and cellular injury. Release of mitochondrial content is mainly driven by BAX and BAK1, which have pro-apoptotic properties. The extrinsic apoptotic pathway is initiated by death receptors (TNFR1, for example) located on the cell membrane. If caspase-8 is present, it is bound by the complex formed by RIP1 and FADD to form complex IIa, leading to apoptosis. Ultimately, both pathways culminate with the activation of caspase-3 (and also of the “executioner” caspases -6 and -7). Both the intrinsic and the extrinsic pathways can be triggered by the loss of integrin anchorage to the extracellular matrix. In this latter instance the apoptotic process is referred to as anoikis. ECM, extracellular matrix.
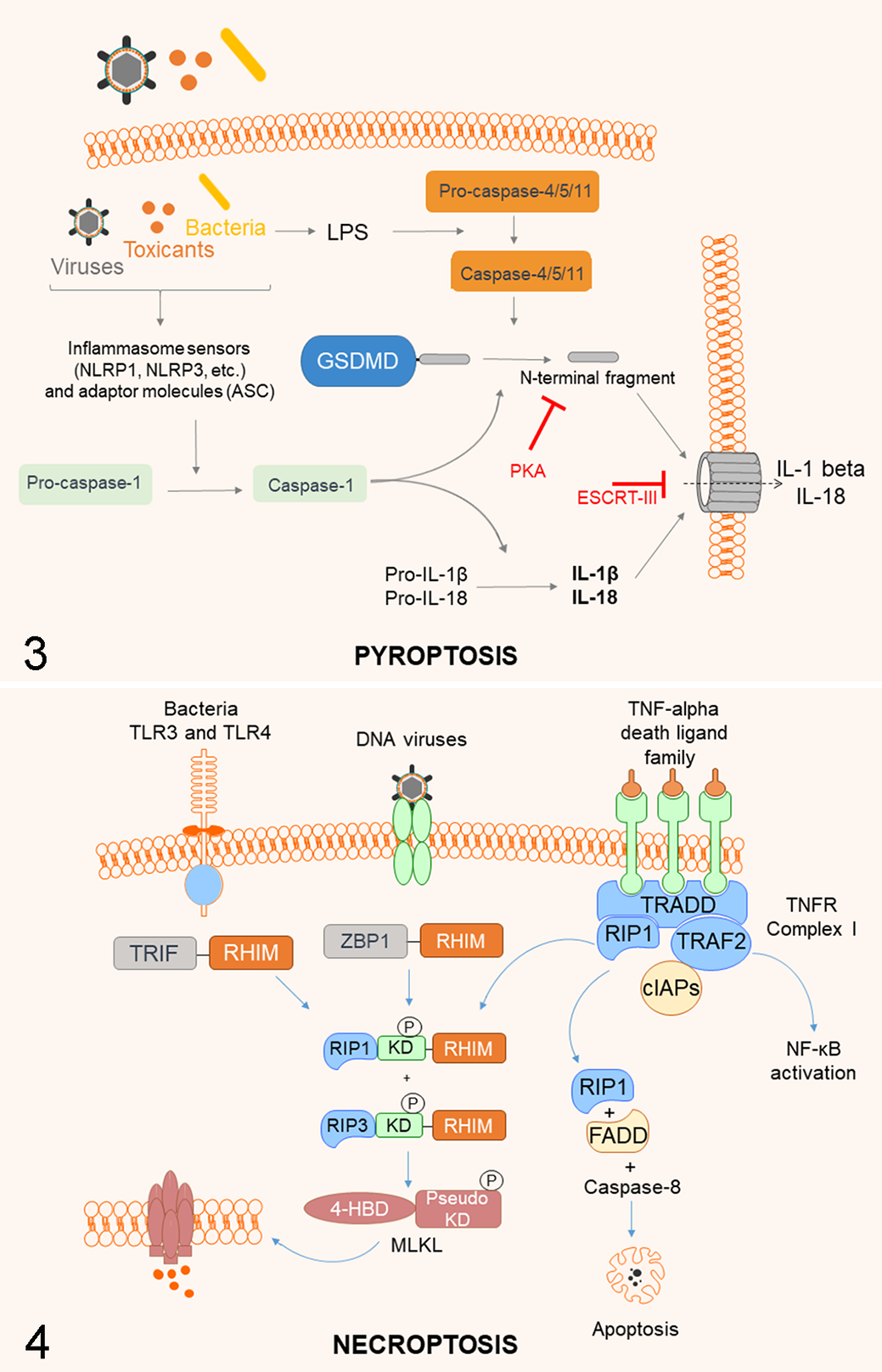
Pyroptosis. In the canonical (caspase-1-dependent) pathway, viruses, bacteria, toxicants, or endogenous proteins (damage-associated molecular patterns, DAMPs) induce the activation of the NLRP3 or NLRP1 inflammasome, which actives pro-caspase-1 to caspase-1. Caspase-1 cleaves the precursors of pro-inflammatory cytokines IL-1beta and IL-18 and gasdermin D (GSDMD). The cleaved N-terminal fragments of GSDMD oligomerize to form the transmembrane pore, allowing for rapid cell membrane permeabilization and leakage of cytoplasmic content including IL-1beta and IL-18. Alternatively, in the noncanonical (caspase-1-independent) pathway, bacterial lipopolysaccharides (LPS) directly activates caspase-4/-5 (or murine equivalent caspase-11), which then cleave GSDMD and initiate pyroptotic cell death. Negative regulators of pyroptosis include PKA that blocks the production of the N-terminal GSDMD fragment, and ESCRT-III that triggers cell membrane repair upon GSDMD activation.
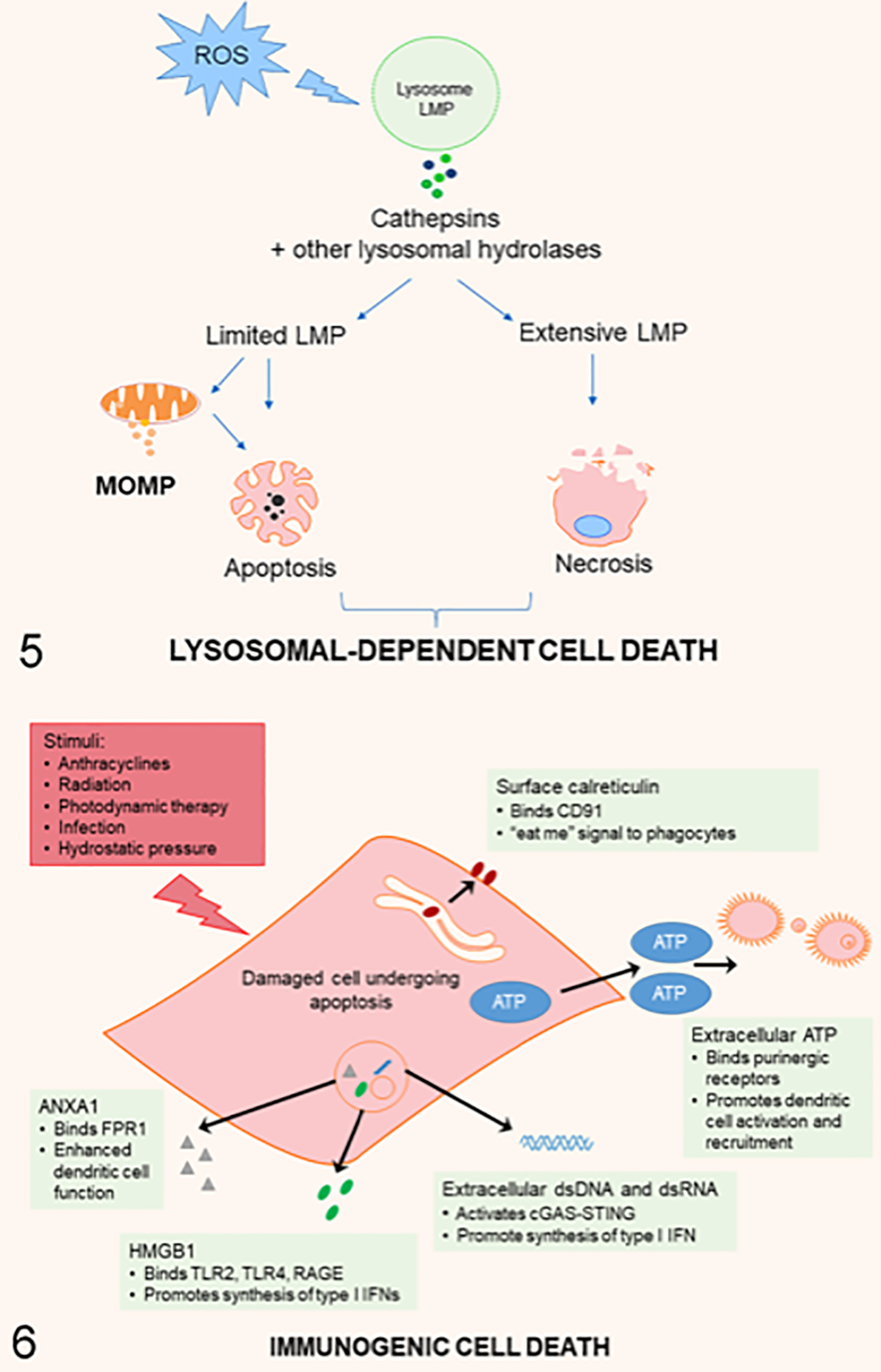
Lysosomal-dependent cell death (LDCD). Classic ROS (reactive oxygen species)-mediated mechanism of lysosomal membrane permeabilization (LMP) leading to LDCD. ROS-dependent LMP leads to translocation of cathepsins from the lysosomes to the cytoplasm, resulting in mitochondrial outer membrane permeabilization (MOMP) with cytochrome c release, caspase activation, and apoptosis. Extensive release of lysosomal enzymes induces necrotic cell death. Permeabilization of the lysosomal membrane can also occur downstream of MOMP.
Experimentally induced cerebral ischemia/reperfusion injury, brain, pig. Acute neuronal death in the CA2 region of the hippocampus is characterized by hypereosinophilic cytoplasm and pyknosis. Hematoxylin and eosin (HE). Inset: dead neurons have intense green fluorescence when labeled with Fluoro Jade C (FJ-C). Neuronal death resulting from cerebral ischemia/reperfusion injury has been associated with ferroptosis, mitochondrial permeability transition-driven cell death, and necroptosis. Courtesy of Prof Eugenio Scanziani, University of Milan.
Anoikis, from the Greek word meaning “home” or “homelessness,” is a specific form of apoptosis (Figs. 1, 9) associated with the loss of interaction or attachment of the cell to its neighboring extracellular matrix (ECM). 234 It plays a crucial role in the prevention of adherent-independent cell growth and of the adhesion of cells to inappropriate ECM proteins. 114,234 The first description of anoikis dates to the early 1990s with the description of cells undergoing classical apoptosis when lacking proper attachment to their ECM. 92,114,211 These initial descriptions of anoikis introduced essential aspects of this form of apoptosis, including the importance of surface receptors of the integrin family, the inhibitory effect of BCL2 on anoikis, and the increased sensitivity to anoikis of certain cell types (eg, epithelial and endothelial cells) as compared to others (eg, mesenchymal cells such as fibroblasts). 92,114,211
Different cell types express diverse sets of integrins that interact with different ECM proteins. Furthermore, the protective effect of integrins is often combined with the effects of growth factor receptors (GFRs). For example, mammary epithelial cells depend on adhesion to laminin via integrin α6β1 along with the activation of the insulin-like growth factor 1 (IGF1) signaling cascade. 86,114,184 Attachment to laminin appears to be a requirement for IGF1 receptor signaling. In fact, the same cells grown on collagen I undergo anoikis, with or without IGF1. 86,114,184
Questions remain open around the initial trigger of the anoikis cascade and the actual attachment sensor aside from the central role played by integrins. 199 Due to the role of integrins as mechanotransducers, anoikis may be viewed as the outcome of a mechanosensory event in which the attachment sites, preferentially integrin complexes, fail to support an internal contractile force due to loss of external anchorage. After loss of integrin-dependent anchorage and detachment of the cell from its ECM, or adhesion to an inappropriate type of ECM, both the intrinsic and the extrinsic apoptotic pathways can be initiated and result in anoikis. 114,122 The choice of mechanisms that regulate anoikis varies depending on cell type, with different integrins activating distinct signaling cascades and apoptotic pathways. 114 In human keratinocytes and umbilical vein endothelial cells, there is upregulation of Fas ligand (FasL) and Fas receptor after cell detachment, alongside the downregulation of Flice-like inhibitory protein (FLIP), an endogenous antagonist of caspase-8; these processes lead to caspase-8 activation and extrinsic apoptosis. 6,204 In mammary epithelial cells, accumulation of the pro-apoptotic factor BIM induces the oligomerization of BAX and BAK1 in the outer mitochondrial membrane, leading to intrinsic apoptosis upon loss of integrin-mediated adhesion. 234 In addition to the canonical apoptotic pathways, unligated integrins can also act as cell-death promoters through a process named integrin-mediated death (IMD). IMD has been described in epithelial cells, for both integrins β1 and β3; it involves the recruitment of caspase-8 to the integrin β subunit cytoplasmic domain and the onset of an apoptotic response. 196,347 A similar direct interaction of unligated integrins and caspase-3 has been described in vitro. 241
In physiological conditions, anoikis is required for normal cellular function such as the turnover of enterocytes or other labile tissues lining different organs, 123 and contributes to developmental processes such as the hollowing of glands. 60 In diseases, the critical role of anoikis is probably best understood in the context of tumor invasion and metastasis. 234 Resistance to anoikis is a hallmark of metastatic malignancies allowing anchorage-independent growth and survival during tumor dissemination. 33 Numerous studies have suggested that stimulation of pro-survival signals and suppression of death signals are involved in anoikis resistance. Neoplastic cells can adapt to the loss of anchorage and avoid anoikis through various ways, including switching their integrin expression, 22,150,234,242 activating IGF1 receptor (IGFR1) and EGFR (epidermal growth factor receptor) family members, activating anti-apoptotic pathways such as phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) 3,234,288 and extracellular signal-regulated kinase (ERK)-mediated suppression of BIM, 33 or shifting to a mesenchymal phenotype (epithelial-mesenchymal transition [EMT]) leading to epigenetic changes conferring resistance to anoikis. 234
All these changes, in combination with modifications in the neoplastic cells’ energy metabolism 234 and transcription factors such as hypoxia-inducible factors (HIFs), 166,289 allow them to survive and thrive in suboptimal conditions.
Ferroptosis
Ferroptosis is a recently recognized RCD that relies on a mechanism of iron-dependent cell injury (hence the term ferroptosis) accompanied by a dysfunction of the cellular antioxidant defense system. The activation of ferroptosis ultimately results in uncontrolled oxidative stress with accumulation of lethal levels of lipid peroxides and eventual cell death (Fig. 2).
First described as a novel RCD modality in 2012, 69 and named after the strict iron dependency of the process, Dixon and colleagues unraveled the mechanism behind erastin-induced ferroptosis via the inhibition of system xc- and the depletion of intracellular glutathione (GSH). GSH is important for the cellular antioxidant defense system, either directly by interacting with reactive oxygen/nitrogen species (ROS and RNS) and electrophiles or by operating as a cofactor for various antioxidant enzymes. It was later elucidated that the key role played by GSH in controlling ferroptosis is attributable to its function as a cofactor of glutathione peroxidase 4 (GPX4). GPX4 protects cells against ferroptosis by arresting the peroxidation of cell membranes. Later it was shown that the presence of free intracellular iron and lipid peroxides are both prerequisites for the execution of ferroptosis. In this context, iron chelators (eg, deferoxamine) as well as lipophilic antioxidants (eg, liproxstatin-1, vitamin E, and nicotinamide adenine dinucleotide phosphate [NADPH]) proved to be potent inhibitors of ferroptosis. 69,158 More recently, GSH-independent forms of ferroptosis, resulting from the dysfunction of the mevalonate pathway and ferroptosis suppressor protein 1 (FSP1), have also been elucidated. 73
Ferroptosis can be triggered by a plethora of stimuli and conditions that ultimately results in unrestrained lipid peroxidation leading to a failure of integrity of the cell membrane or membranes in subcellular locations (ie, organelles).
Much of what is known today about ferroptosis comes from in vitro experiments 52 and the in vivo relevance of this process is still largely unexplored. 72 Nonetheless, there is preliminary evidence that ferroptosis may be implicated in a wide range of pathologic processes including ischemia/reperfusion injury, CNS trauma, amyotrophic lateral sclerosis, hemochromatosis, disorders of spermatogenesis, and doxorubicin toxicity. 30,66,190,280,309,325,329 Ferroptosis also provides a possible explanation for the “metal hypothesis” of Alzheimer’s disease and other neurodegenerative disorders, 207 which proposes that the neuropathogenic effects of amyloid-beta (Aβ, a peptide thought to be neurotoxic) are promoted by and possibly dependent on the interactions between metals and Aβ. 34 Tissue changes observed in the above-mentioned conditions do not have morphological findings that can be considered typical for ferroptosis. Nevertheless, the combination of ultrastructural features of cells undergoing ferroptosis, at least in vitro, are distinct from the ones observed during other forms of RCD. These include cell rounding, cell membrane rupture, smaller mitochondria, higher mitochondrial membrane density, vanishing of mitochondrial cristae, and the rupture of the mitochondrial outer membrane. 187,284
Ferroptosis has become an intense area of investigation in cancer research as an alternative way to eradicate cancer cells that escape other forms of cell death or are resistant to conventional therapy. 13 The availability of several small molecules capable of inducing ferroptosis (eg, erastin, sorafenib, FINO2), some of which are approved by the US Food and Drug Administration (FDA) for clinical use, has already provided promising nonclinical evidence of efficacy against specific types of cancers including non–small cell lung cancer, hepatocellular carcinoma, diffuse large B cell lymphoma, and renal cell carcinoma. 129,219,226,324
NETosis
NETosis is a distinct RCD pathway that occurs primarily in neutrophils. NETotic cell death leads to the release of neutrophil extracellular traps (NETs, hence the term NETosis). NETs are decondensed chromatin structures with antimicrobial properties that can trap and kill various bacterial, fungal, and protozoal pathogens. 93 Neutrophils “cast” NETs particularly in response to large microbial structures that cannot be easily phagocytosed such as Candida albicans hyphae and Mycobacterium bovis aggregates. 25
In 1996, Takei and colleagues uncovered the first evidence of a neutrophil-specific type of cell death triggered by phorbol myristate acetate (PMA), a potent activator of neutrophils. 281 This process would later be referred to as NETosis following the description and characterization of the extracellular NETs released by dying neutrophils upon stimulation with interleukin (IL)-8, PMA, or lipopolysaccharide (LPS). 29 It was eventually discovered that other cells of the innate immune system, such as eosinophils, basophils, and macrophages, may occasionally activate a similar mechanism of cell death referred to as ETosis. 118,236
Similar to phagocytosis and degranulation, NETosis is a component of the neutrophil arsenal and part of the first line of defense against pathogens. 29 During NETosis, the neutrophil extrudes tentacles of chromatin armed with antimicrobial proteins forming a viscous web that entraps and kills pathogens. 162 NETosis can be initiated by a variety of stimuli, including microbial structures such as LPS and flagella, 29 immune complexes, 169,181 complement activation products, 185 damage-associated molecular patterns (DAMPs), 327 proinflammatory cytokines, and activated platelets. 50 Different receptors, such as Toll-like receptors (TLRs), Fc receptors, or complement receptors, have been reported to sense these stimuli and mediate NETosis. 162 In primed neutrophils, NETosis starts with the release and activation of several mediators contained in neutrophil granules including neutrophil elastase (NE), myeloperoxidase (MPO), matrix metalloproteinases (MMP), and the NADPH oxidase. 29,273 While NADPH oxidase and MPO lead to the massive production of ROS, NE contributes to the proteolysis of histones and decondensation of nucleosomes. 28,162 High levels of ROS activate the protein-arginine deaminase 4 (PAD4), which plays a major role in the chromatin decondensation process by mediating the hypercitrullination of histones, a posttranslational modification that leads to NET formation. 162,317 The intracellular oxidative stress that follows the initiation of NETosis results in extensive damage and dissolution of cellular membranes allowing the decondensed chromatin to mix with cytosolic microbicidal proteins released by neutrophil granules, including cathepsin G, NE, MPO, gelatinase, lysozyme, calprotectin, defensins, and cathelicidins such as the cathelicidin antimicrobial peptide (CAMP). 29 These components are complexed with the chromatin constituting the weaponry that kills pathogens when NETs are extruded extracellularly. The whole process of NET formation is completed 1 to 4 hours after the inciting stimulus and is often lethal to neutrophils. 29,162 The activation of the cyclin-dependent kinases 4 and 6 (CDK4/6) is critical for the release of NETs. 4
Aside from this suicidal NETosis requiring membrane rupture and the loss of conventional live neutrophil functions, a nonsuicidal pathway of NETosis called vital NETosis allows NET release and conventional host defense to coexist. 334,335 In vivo, NETs are degraded by plasma DNases and subsequently cleared by phagocytes. 28 Defective NETs clearance represents a possible trigger of an autoimmune response against intracellular components that are associated with the extruded chromatin projections. 28 In this context, the development of autoimmune diseases like systemic lupus erythematosus has been linked to the prolonged or persistent extracellular exposure of histone complexes resulting from impaired NET degradation. 126
An increasing body of evidence, in both human and veterinary medicine, suggests that, depending on the pathophysiological context, NETosis may have protective or detrimental clinical repercussions. For example, during bacterial infections, NETs protect the host by limiting microbial growth and dissemination especially via immunothrombosis, a process in which an innate immune response induced by the formation of thrombi facilitates the recognition of pathogens and damaged cells. 82,117,189,275 However, unregulated activation of NETosis contributes to serious medical complications characterized by generalized circulatory disorders. Recent clinical evidence underpins the crucial role of NETs in the pathogenesis of intravascular thrombosis, disseminated intravascular coagulation (DIC), and multiple organ dysfunction, all of which can increase morbidity and mortality during sepsis. The complex interactions existing between activated platelets, neutrophils, and NET formation have been recognized as one of the critical determinants promoting dysregulated immunothrombosis and subsequent circulatory disorders. 117
In addition to playing a central role in the context of sepsis across numerous mammalian species, NETosis has been implicated in specific diseases of veterinary interest including intestinal coccidiosis in ruminants, 220,270 bacterial metritis in mares, 244 immune-mediated hemolytic anemia in dogs, 183 and Mannheimia haemolytica pneumonia in cattle (Fig. 10). 10 In this latter example, a specific bacterial leukotoxin is responsible for the massive activation of NETosis, and the streaming of the extracellular chromatin extruded by NETotic bovine neutrophils is so prominent that it assumes a peculiar histopathological pattern known as “oat cells.” 10 NETs are also generated during viral infections. While NETs can contribute to control of the infection by trapping and inactivating the viral particles, 137 disproportionate virus-induced NET release may promote local tissue damage as well as serious systemic repercussions. In this case, endothelial cell damage, formation of autoantibodies, and deposition of immune complexes through stimulation of self-reactive memory B cells can lead to multi-organ damage and promote autoimmune processes. 258 In cats infected with feline leukemia virus where NETosis is overactivated, disease progression is promoted especially in response to secondary infection. 125
Parthanatos
Parthanatos is a recently recognized form of RCD that occurs in response to extreme genomic stress with extensive DNA damage. 58 Parthanatos is associated with the overactivation of the enzyme poly(ADP-ribose) polymerase-1 (PARP1), a key member of the DNA repair machinery. 58 Cell death resulting from PARP1 overactivation with consequent release of apoptosis-inducing factor mitochondria associated 1 (AIFM1; best known as apoptosis-inducing factor [AIF]) from the mitochondria was first demonstrated by Yu and colleagues in 2002. 339 However, it was only in 2009 that the term parthanatos was coined after Thanatos, the personification of death in Greek mythology, to describe cell death initiated by poly(ADP-ribose) (PAR). 58 More recently, in 2016, Wang et al screened AIF-binding proteins and identified the macrophage migration inhibitory factor (MIF) as the effector nuclease that carries out the final DNA destruction step of parthanatos. 315
Parthanatos features a specific cell death modality resulting from the execution of a well-orchestrated cascade of events that is distinct from apoptosis, although some analogies exist in terms of molecular, morphological, and structural changes. 87 Sustained DNA damage caused by genotoxic stress or excitotoxicity represent the main inciting stimulus for the activation of parthanatos. The pathway is characterized by key distinguishing sequential steps, including the rapid activation of PARP1, and synthesis and accumulation of the PAR polymer. 151,315 The increasing accumulation of PAR triggers AIF release from the mitochondria. AIF then translocates to the nucleus, recruiting MIF nuclease from the cytosol and activating it to produce large-scale DNA fragmentation (≈50 kb, as opposed to the small-scale DNA laddering seen in apoptosis), chromatin condensation, and, eventually, cell death. Parthanatos is therefore a caspase-independent cell death modality that requires the nuclease activity of MIF to execute the “deadly blow” characterized by DNA destruction. 151 Unregulated accumulation of PAR has been identified as a key event in the activation of the molecular cascade that ultimately leads to parthanatos. Cellular levels of PAR are regulated in the cell by the enzyme poly(ADP-ribose) glycohydrolase (PARG), which catalyzes PAR degradation, and by ADP-ribosyl-acceptor hydrolase 3 (ARH3), an enzyme with PARG-like activity. 208 In this context, PARG has been found to protect against parthanatos, while its deletion results in increased cell death through the accumulation of PAR. 58,339 In addition, the PAR-dependent E3 ubiquitin ligase ring finger protein 146 (RFN146, also known as IDUNA) was shown to bind to cytosolic PAR chains and prevent PARP1-induced AIF release and parthanatos. 5
There are several well-known pathological conditions whose pathogenesis involves PARP1 overactivation and possibly parthanatos. The CNS appears to be particularly sensitive to this type of RCD modality, especially neurons. It was in the CNS that PARP1-dependent cell death was first detected in response to RNS insult. 345 Following this seminal discovery, involvement of PARP1 overactivation has been demonstrated to have a role in experimental models of excitotoxicity, post-ischemic brain damage, 78 trauma, 182 Alzheimer’s disease, 197 Huntington’s disease, 301 amyotrophic lateral sclerosis, 139 and spinal cord injury. 200 Notably, PARP1 activity has been linked to the neurodegenerative properties of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinsonism in 129S/SvEv 203 and C57BL/6 56,336 mice. In addition, in the neurons of individuals with Parkinson’s disease, formation of pathologic alpha-synuclein deposits in the form of Lewy bodies is associated with activation of PARP1 and increased PAR. 159 Outside the CNS, parthanatos has been experimentally linked to renal cadmium toxicity in rats. 198
Mitochondrial Permeability Transition (MPT)-Driven RCD
The role of mitochondria in RCD is well known and has been extensively studied. 102,231 The intrinsic apoptotic pathway is initiated by mitochondrial permeabilization that results in caspase activation. 102 Mitochondria are also essential in MPT-driven necrosis, a form of RCD caused by a disturbance of the intracellular microenvironment and characterized by a necrotic phenotype. The best-known manifestation of MPT-driven necrosis is seen in various organs after transient loss of vascular patency, leading to ischemia/reperfusion injury (eg, cerebral and myocardial infarctions). 59,217,255 Severe oxidative stress and intracellular calcium (Ca2+) overload induce the opening of the permeability transition pore complex (PTPC) located between the inner and outer mitochondrial membranes. 105,217,231 This results in an abrupt increase in inner mitochondrial membrane permeability, influx of solutes and water in the mitochondrial matrix, loss of mitochondrial transmembrane potential, and depletion of ATP, ultimately leading to rupture of the outer mitochondrial membrane and release of mitochondrial enzymes culminating in the cell death. 102
While not part of the pore-forming unit of the PTPC, the cyclophilin D protein (CYPD) is the only protein that has been proven to be essential in MPT-driven necrosis. 12,223 Confirming the essential role of CYPD, CYPD inhibitors such as cyclosporin A, sanglifehrin A, and JW47 reduced the cellular effects (eg, mitochondrial swelling and cell death) and improved the functional/clinical outcomes (eg, altered left ventricular pressure and loss of motor functions) of MPT-driven necrosis in rodent models of ischemia/reperfusion injury in various organs including heart and brain. 12,51,255,319 Furthermore, HAX1 (HCLS1-associated protein X-1) negatively regulates MPT-driven necrosis through degradation of CYPD and limits the demise of mouse cardiomyocytes in ischemia/reperfusion models. 180 The exact composition of the PTPC still remains elusive, although a number of other proteins have now been shown to play a physical or functional role in the PTPC and thus in MPT-driven necrosis, 105 such as members of the BCL2 family (including BID, 341 BAX, 321 BAK, 163 BCL2, and BCL-XL), 296 tumor protein 53 (TP53; when physically associated with CYPD), 299 F-ATP synthase, 115 and others. 148 The complexity surrounding BCL2 family members derives from their role in mediating not only apoptosis but also a number of apoptosis-unrelated functions to maintain homeostasis. 48 In MPT-driven necrosis, the mitochondrial pools of BCL2, BCL-XL, and MCL1 interact with numerous members of the voltage-dependent anion channel (VDAC) protein family that regulates fluxes of ions across the outer mitochondrial membrane promoting supra-physiological concentrations of Ca2+ within mitochondria. However, whether BCL2 proteins favor VDAC opening or closure to drive MPT is still debated. 48
Whether necrosis or intrinsic apoptosis occurs following mitochondrial permeability seems to be context-dependent and still largely unclear. Studies conducted on experimental models of myocardial infarction and stroke have suggested that the apoptotic or necrotic outcome might be MPT threshold-dependent. In this context, modest increase in mitochondrial permeability results in apoptotic cell death, whereas severe impairment of mitochondrial membrane homeostasis leads to a necrotic phenotype. 141
Pyroptosis
Pyroptosis (Figs. 3, 11), also called caspase-1-dependent cell death, plays a crucial role in the innate immune system for the defense against microbial infections. 15,152,266 The term pyroptosis was first proposed in 2001 by Cookson and Brennan, 55 from the Greek pyro, fire or fever, due to its remarkable pro-inflammatory properties. However, the first mention of what is now known to be pyroptosis dates to 1986 in relation to the cytotoxic effects of anthrax lethal toxin on murine peritoneal macrophages. 91,266 Similar observations were later evidenced with macrophages exposed to Shigella flexneri 352 and Salmonella typhimurium 21,27 and were noted to be caspase-1-dependent.
Originally believed to be limited to monocytes and macrophages, pyroptosis has since been described in various cell types in response to bacterial insults such as LPS (Fig. 3). 164,165,171,260,268 As opposed to apoptosis and other mechanisms of cellular death, pyroptosis is associated with the development of an inflammatory response.
The critical protective role played by inflammatory caspases and pyroptosis against a variety of bacterial pathogens has been mainly inferred from experimental studies in genetically manipulated mice. 202 However, given its inextricable role in infectious diseases, pyroptosis has been also recently implicated in numerous diseases of domestic animals, such as porcine reproductive and respiratory syndrome virus (PRRSV) infection and classical swine fever virus infection, in which it causes release of IL-1beta by macrophages. 305 The finding of increased NLRP3 labeling in the glomeruli of dogs infected with Leishmania infantum led to the hypothesis regarding a potential role of the NLRP3 inflammasome engagement in the pathogenesis. 83 Although little is known about inflammasome responses in cattle, in vitro studies demonstrated caspase-1 activation in bovine herpesvirus 1 infections, as well as IL-1beta secretion from bovine monocyte-derived macrophages in the presence of bovine viral diarrhea virus type 2 and LPS. In addition, breed-specific differences in inflammasome-related responses in cattle exist. 305 In natural hosts of anthrax, such as cattle and bison, the NLRP1 sequence is not susceptible to cleavage by lethal toxin (LT), and their macrophages are resistant to toxin-induced rapid cell death and only undergo a slower non-pyroptotic death when the LT is present. 304 Last, Bartonella tribocorum has been shown to evade intracellular destruction by inhibiting pyroptosis of macrophages, which allows the bacteria to survive and travel through the lymphatic system from the site of infection. 142
Lysosome-Dependent Cell Death
Lysosomes function as the cellular recycling center. They are filled with several hydrolases that can break down most cellular macromolecules. Lysosomal membrane permeabilization along with the subsequent leakage of the lysosomal contents into the cytosol leads to so-called lysosomal cell death or lysosome-dependent cell death (LDCD; Fig. 5). 1
LDCD is mainly carried out by proteases of the cathepsin family. It is important to mention that the outcome of lysosomal membrane permeabilization is directly dependent on multiple factors including the rate of lysosome disruption, the type of stimulus, the type of cathepsins released into the cytoplasm, and the presence of cathepsin inhibitors. 1,24 When the disruption of lysosomes is extensive or complete, the rapid increase in cytosolic acidity and leakage of lysosomal enzymes lead to uncontrolled breakdown of cell components, resulting in cell death with the typical morphological features of necrosis. 308 In contrast, limited and selective permeabilization of the lysosomal membrane induces cell death through the intrinsic apoptotic pathway in apoptosis-competent cells 155 or through a caspase-independent cell death pathway with apoptosis-like morphology in cells with defective apoptosis. 170
The ability of microinjected cathepsin B or D to trigger MOMP and apoptosis, 20,246 along with the ability of cathepsin B to induce apoptotic morphology in isolated nuclei, 295 supported the role of cathepsins as executors of LDCD. This is further reinforced by the significant protection against cell death following the inhibition of cathepsins, either by genetic or pharmacological targeting, or by overexpression of cytosolic cathepsin inhibitors. 1 However, while cathepsins are vital executors of LDCD, it should be emphasized that cathepsin inhibition only provides partial protection from LDCD, indicating the yet-unidentified roles of other lysosomal hydrolases (eg, lipases and phosphatases) and lysosomal membrane permeabilization-associated dysfunction in LDCD. 1
LDCD is relevant for several pathophysiological conditions including inflammation, tissue remodeling, aging, neurodegeneration, cardiovascular disorders, and intracellular pathogen response. 1,23,119,261 During postlactation mammary gland involution, signal transducer and activator of transcription (STAT) 3 has been shown to promote LDCD by upregulating the expression of cathepsins while downregulating their endogenous inhibitor Serpina3g (serine protease inhibitor A3G; best known as serine protease inhibitor 2A [Spi2A]). 174,252 Currently, very little is known about the relevance of LDCD in diseases of animals. Bosutinib, a second-generation tyrosine kinase inhibitor, induced lysosomal membrane permeabilization, which resulted in the release of lysosomal hydrolases including cathepsin B and subsequent caspase-independent cell death in canine melanoma cells. 227 In a rat model of glioma, the intracerebral injection of H1-parvovirus induced tumor regression associated with increased activity of cathepsin B and cytosolic reduction of its inhibitors cystatin B and C. 68
While LDCD is the most common cell death pathway in which lysosomes are implicated, in certain conditions, the presence of lysosomal damage, membrane permeabilization, and intracytoplasmic release of lysosomal content ultimately result in other types of cell death such as pyroptosis or ferroptosis. 308
Immunogenic Cell Death
Immunogenic cell death (ICD) is defined as a “type of RCD that is sufficient to activate an adaptive immune response in immunocompetent hosts.” 105 In ICD, the inflammatory response may culminate with the activation of cytotoxic T lymphocyte-driven adaptive immunity toward endogenous or exogenous antigens expressed by targeted cells (Fig. 6). 101,105,106 Exogenous antigens may be derived from viruses or bacteria. Endogenous antigens are typically neo-epitopes encoded by genes that have mutated during oncogenesis and tumor progression, 101,168 or antigens associated with auto-immune diseases. 106 ICD is initiated by cellular stressors including infections, 112 therapeutic oncolytic viruses, 31,138 some chemotherapeutic agents such as anthracyclines, 37,40,76 some DNA-damaging agents, 287 specific forms of radiation therapy, 67,107 hypericin-based photodynamic therapy, 109 and high hydrostatic pressure, 95 among others. Furthermore, ICD mediates the abscopal effect of radiation therapy, where irradiation of a malignant lesion results in the regression or stabilization of a distant, non-irradiated lesions. 101
Although antigens, including neo-epitopes, are released in many forms of RCD, the hallmark immunogenicity of ICD is conferred by the actions of adjuvants, typically DAMPs that are simultaneously released by the dying cell. Indeed, non-immunogenic forms of cell death can be made immunogenic by boosting the availability of DAMPs. 18 At least 6 DAMPs have been mechanistically linked to ICD: calreticulin (CRT), 108,228 ATP, 79,113,213 high-mobility group box 1 (HMGB1), 7,53,254 type I IFN, 146,272,298 cancer cell-derived nucleic acids, 45,110 and annexin A1 (ANXA1). 294 These DAMPs activate PRRs on cells of the immune system to modulate the immune response toward epitopes exposed through the process of ICD. For example, CRT translocates from the endoplasmic reticulum (ER) to the outer leaflet of the cell membrane due to increased ER stress through the course of ICD. 108,228 CRT on the outer cell membrane functions both as an “eat me” signal for antigen-presenting cells (APCs) and as a trigger for Th17 priming. 235,239 The other DAMPs implicated in ICD similarly function to promote recruitment and activation of APCs, phagocytosis, and the synthesis of pro-inflammatory cytokines. 101 Of note, ICD is robustly suppressed by caspase-3 and -8 through various mechanisms. 103
Enhancing the immunogenicity of tumor cells is an active area of research within the field of immuno-oncology in humans and animals, including radiation therapy applied to canine oral malignant melanoma 143 and various strategies involving autologous vaccination. 333
Entosis
The actomyosin-dependent cell-in-cell internalization (entosis) represents a form of cell cannibalism that is followed by execution or cell killing 191 by lysosomes through the lysosomal degradation pathway (ie, entotic cell death). 105,283 Entosis was initially described as a nonapoptotic cell death process triggered by epithelial cells in the absence of attachment to the basement membrane or ECM and consequent loss of integrin signaling. 209,233 In contrast to anoikis, which is triggered on adhesion loss and is characterized by caspase activation, the hallmark of entosis is the formation of cell-in-cell structures through an invasion mechanism in which cells actively drive their own uptake into neighboring cells after formation of cell-cell adhesions. 84 This process is most commonly followed by the death of the internalized entotic cells by their hosts following maturation of the endocytic membrane. 90,233
Entosis has been mostly reported in tumors of epithelial origin undergoing aberrant cell proliferation and matrix detachment. 233 Alternative mechanisms include metabolic stress induced by glucose starvation in proliferating cancer cells competing for resources; 127 deregulation of myosin activation during cell-cell contact formation without matrix detachment; 307 and differences in the mechanical properties between the engulfing and the engulfed cells. 277 A form of entosis provoked by mitotic stress (entotic mitosis) driven by dramatically increased mitotic rounding (ie, increased cortical rigidity of the cells entering mitosis) upon depletion of cell division cycle 42 (CDC42) or exposure to antimitotic agents has been also described. 77
Both cell adhesion and cytoskeletal rearrangement play a central role in the control of entosis induction. 206 Entosis requires the formation of junctions between engulfing and engulfed (entotic) cells mediated by adhesion proteins cadherin 1 (CDH1; also known as E-cadherin) and catenin alpha 1 (CTNNA1) without the involvement of integrin receptors. 233,277,313 Following cadherin-mediated homotypic interactions between neighboring sibling cells (ie, cadherin-mediated adhesive interaction with the same cadherin species expressed by neighboring cells), the cytoplasmic tail of E-cadherin binds to p120 and beta-catenin intracellularly; in turn, beta-catenin links E-cadherin to the actin cytoskeleton via an interaction with alpha-catenin. 300 The accumulation of actomyosin chains is essential for the formation of cell-in-cell structures, also described as “bird’s eye cells” (Fig. 12), which represent the main morphological feature of entosis. 206
This mechanism is controlled by the ras homolog family member A (RHOA), Rho-associated coiled-coil-containing protein kinase 1 (ROCK1), ROCK2, and protein diaphanous homolog 1 (DIAPH1). 175 The result is a contraction of cytoskeletal elements that promotes engulfment. 233,277,313 Other signaling molecules and regulators of entosis include aurora A kinase (AURKA) and AMP-activated protein kinase (AMPK) through modulation of microtubule plasticity or energy metabolism; 323 CDC42, whose depletion enhances changes in mitotic morphology; 77 and chromatin-binding protein nuclear protein 1 (NUPR1), which negatively regulates entosis through modulation of AURKA activity or autophagy. 136 The ultimate digestion of the engulfed cells requires uptake by BCL2-independent microtubule-associated protein 1 light chain 3 beta (LC3)-associated phagocytosis (LAP), and degradation by a cathepsin B-mediated lysosomal pathway. 77,206 In this context, some components of the macroautophagy apparatus such as LC3, autophagy-related 5 (ATG5), ATG7, PI3K catalytic subunit type 3, PI3K regulatory subunit 4, and beclin-1 (BECN-1) promote the fusion of entotic cell-containing vesicles with lysosomes. 90
Despite the fact that characterization of entotic death is still in its infancy, cell-in-cell structures have been documented in several human neoplasms. 233 However, the role of entosis in tumorigenesis remains ambiguous, 105,283 as both tumor suppressive mechanisms 233,307 and promotion of tumor progression have been reported. 149,302,303 Thus, the clinical relevance of cell-in-cell structures remain largely unknown. 90 Physiological roles of entosis have been described for uterine luminal epithelial cells during embryo implantation to allow trophoblast cells to contact the underlying stroma. In fact, implantation was impaired in mice treated with a ROCK inhibitor. 191
To date, the implications of entosis in spontaneous animal diseases are unknown. Furthermore, it is still uncertain whether this phenomenon could be used for therapeutics applications.
Necroptosis
Necroptosis represents a regulated, pro-inflammatory, caspase-independent form of necrotic cell death. This term was introduced in 2005 to indicate cell death in ischemic neuronal injury triggered by TNFR1 in the presence of caspase inhibition. In this context, it was demonstrated that this cell death process is dependent on receptor-interacting serine/threonine kinase 1 (RIPK1) and can be inhibited by necrostatin 1 (Nec-1). 62,63 Subsequently, necroptosis was better defined as a RIPK3-dependent molecular cascade promoting regulated necrosis 218 based on the discovery of a close interaction between RIPK1 and its homolog RIPK3 in transducing pro-necrotic signals, 47,134,344 and on the identification of multiple RIPK3-dependent but RIPK1-independent instances of regulated necrosis such as in murine cytomegalovirus infections, nitric oxide (NO)-induced pancreatic beta-cell death, and cell death in fibroblasts. 156,282,291,297 This understanding was further enhanced by the discovery of mixed lineage kinase domain-like pseudokinase (MLKL) as the effector of necroptosis (Figs. 4, 13). 276,349
The best-studied form of necroptotic cell death is initiated by TNF-alpha. 2 However, necroptosis can also be induced by other members of the TNF-alpha death ligand family (Fas and TNF-related apoptosis-inducing ligand [TRAIL/Apo2L]), 62 interferons, TLRs (such as TLR3 and TLR4) signaling, viral infection via the nucleic acid sensor DAI (DNA-dependent activator of interferon regulatory factor, also known as Z-DNA binding protein 1 [ZBP1]), 291,292 retinoic acid receptor responder 3 (RARRES3, also known as retinoid inducible gene 1 [RIG1]), 257 transmembrane protein 173 (TMEM173, also known as stimulator of interferon genes [STING]), 26,42 and ligation of adhesion receptors (CD44, CD11b, CD18, or CD15). 314
While the initial stimulus and signal transduction are shared between apoptosis and necroptosis, the cell fate decision relies on caspase-8. When the TNFR complex IIa forms, Fas-associated death domain (FADD) and pro-caspase-8 are assembled to induce apoptosis. 311 Contrarily, when caspase-8 activation in the TNFR complex IIa is impaired, the TNFR complex IIb/n (ie, necrosome) forms and RIP1 initiates necroptosis through RIPK3 recruitment and activation. 47,134 Signaling pathways involved in RIPK3 activation entail the homotypic interaction of RIP homotypic interaction motif (RHIM) domain-containing receptors, adaptors, and kinases. 2 The RHIM domains of RIPK1 and RIPK3 initiate necroptosis and mediate the formation of large hetero-amyloid signaling complexes called “necrosomes.” 188,216 These amyloid complexes consist of β-sheets and are required for necroptotic signaling and activation. 188,216 Once the necrosome is formed, RIPK1 and RIPK3 engage in a series of autophosphorylation events that are essential for necroptotic cell death. 2 In particular, phosphorylation of RIPK3 allows the binding and phosphorylation of MLKL, prompting MLKL oligomerization, cell membrane permeabilization, and cell rupture. 221,276,310,349
Morphologically, necroptotic cell death manifests with necrotic features including swelling of the cytoplasm and its organelles, early rupture of cell membrane and organelles without formation of apoptotic bodies, and minimal nuclear changes (ie, the changes include chromatin condensation, and nuclear and DNA fragmentation). 2 In necroptosis, the release of cellular components into the extracellular microenvironment triggers inflammation, in contrast to the situation with apoptosis.
During development, the roles of necroptosis include the elimination of potentially defective cells prior to birth and the maintenance of T-cell homeostasis. 105 In cancers, key regulators of the necroptotic pathway are generally downregulated, suggesting that cancer cells may evade necroptosis to survive. 120,132 Necroptotic cell death appears to be involved in various diseases including septic states induced by Gram-negative bacteria, acute pancreatitis, ischemia/reperfusion injury, inflammatory bowel disease, neurodegenerative diseases, traumatic brain and spinal cord injuries, retinal degeneration, and Huntington’s disease. 46,153 Interestingly, while RIPK1 is found in the vertebrate genomes, RIPK3 and MLKL are absent from many genomes. In marsupials (wallaby, opossum, and Tasmanian devil), MLKL and RIPK3 are both absent, implying that necroptosis would not occur. In chickens, RIPK3 is absent, but MLKL is present. On the other hand, carnivores have RIPK3 but lack MLKL. 225 In domestic animals, recent findings suggest a role of necroptosis in steroidogenic luteal cell death and elimination during luteolysis in cows, 140 and in experimental cadmium-induced cardiotoxicity in chickens. 36
Autophagy-Dependent Cell Death
Autophagy-dependent cell death (ADCD) is a form of RCD that is dependent on the cellular machinery of autophagy, 64,98,105 a catabolic homeostatic process that culminates in lysosomal degradation of cytoplasmic materials (Fig 14). 98
Initially, due to the morphologic differences between cells that die as they accumulate autophagosomes within their cytoplasm and cells undergoing apoptosis or necrosis, the terms “autophagic cell death” or “type II programmed cell death” (as opposed to type I cell death, which is identified with apoptosis) were adopted. 100,259 However, the term autophagic cell death has a morphologic connotation and does not imply a causative role of autophagy in the cell death process. Further confusion in the definition of the role of autophagy in the cell death scenario is generated by the use of terms autophagy-associated cell death and autophagy-mediated cell death. 64 According to the NCCD, these two latter designations should be used as modifiers to describe specific cell death modalities, such as apoptosis or ferroptosis, when those are preceded or accompanied by the induction of autophagy. On the other hand, the NCCD defines ADCD as a distinct form of RCD that mechanistically depends on the autophagic machinery without the involvement of other cell death modalities. 64
ADCD describes an instance of cell death occurring due to failure of the cellular autophagic response to preserve homeostasis. 100 Specifically, ADCD is caused by impaired regulation of macroautophagy, a specific subtype of autophagy in which large portions of the cytoplasm, including whole organelles, are degraded in double-membrane autophagosomes. 100,104 As such, ADCD, which mechanistically depends on the autophagic machinery, is prevented by inhibiting autophagy, and lacks involvement of apoptosis or other cell death pathways. 64 This functional definition definitively distinguishes ADCD from other forms of RCD in which the autophagic machinery might be activated but does not play a primary role in driving the cell's demise. 64
Three mechanisms of ADCD have been proposed: excessive autophagy, excessive mitophagy, and autosis. 19 In excessive autophagy, the extreme generation of autophagosomes and overconsumption of cellular organelles result in loss of cellular membrane homeostasis. 57 Cell death due to excessive mitophagy results from accelerated autophagic destruction of mitochondria with failure of cellular energy production. It has been described in various instances of mitochondrial injury including ischemia. 337,351 Autosis is a specific form of ADCD that relies on cell membrane Na+/K+-ATPase. Autosis is induced by autophagy-inducing peptides, such as transactivator of transcription (Tat)-BECN-1 and Tat-viral FLIP-a2, in conditions of cells starvation, and tissue hypoxia-ischemia. 195 Aside from the unique dependence on Na+/K+-ATPase, autosis is characterized by the presence of nuclear membrane changes such as nuclear membrane convolution and shrinkage, focal concavity of the nuclear surface, and focal ballooning of perinuclear space. 194
Besides the activation of the autophagic machinery little is known about the specific biochemical cascades that initiate ADCD. One proposed pathway involves the release of autophagy inhibition, either by upregulation of the pro-autophagic vacuolar protein sorting (Vps34)-BECN-1 complex or sequestration/downregulation of the anti-autophagic beclin-1 inhibitor BCL2. 11,338 In other instances, ADCD may be initiated by noncanonical activation of autophagy independent of the VPS34-BECN-1 complex. 57,351 In addition, mammalian target of rapamycin complex 1 (mTORC1) is suggested to promote growth by negatively regulating the autophagy machinery. 256 Mutations or deletions of essential autophagy-related genes in the fruit fly Drosophila melanogaster result in suppression of normal developmental tissue degradation. 16,65 However, mice with deletions of key autophagy genes develop normally to the neonatal stage, 215 and only exhibit embryonic lethality when apoptotic deficiencies are also induced, 9 suggesting that ADCD is not essential in mammalian development. ADCD may play a role in ischemic injury to the brain and heart. 19 Mice with neuron-specific deletion of Atg7, a gene encoding important autophagy machinery, are resistant to neuronal cell death in a model of neonatal hypoxic/ischemic brain injury. 172,326 Similar protection has been shown in ischemic cerebral injury in rats administered autophagy inhibitors. 89,240 The association between ATG16L1 risk alleles and Crohn’s disease is a well-characterized link to autophagy. 35,249 In this context, ADCD plays a critical role in determining the severity of mucosal damage in rodent models of inflammatory bowel disease. 265 Finally, although autophagy can promote chemotherapy resistance, in some instances ADCD mediates the sensitivity of tumor cells to chemotherapy. 97,263
In domestic animals, changes in the autophagic machinery with accumulation of LC3 in neurites and of the ubiquitin-binding protein p62 in the neuropil of the spinal cord has been demonstrated in Pembroke Welsh Corgi dogs affected by degenerative myelopathy. 230 The accumulation of LC3 and p62 has also been observed in the polyglucosan bodies of dogs affected by Lafora disease. 41 In Lagotto Romagnolo dogs, mutations in the autophagy-related gene ATG4D leads to a progressive neurologic disease linked to altered basal autophagy. 279 However, it is clear that relying solely on morphological features is not sufficient to identify autophagy as the mechanism of cell death. 64
Evaluation of RCD
In the discovery space and in safety assessment, biomarkers and functional tests (including genetic and pharmacological inhibition studies) have been developed to aid in the subclassification of the specific RCD mechanisms and to correlate the expression of designated RCD-specific molecules to known clinicopathological entities. 320 An overview of morphological and biochemical features, pathway regulators, and biomarkers is provided in Table 1 and examples are shown in Figures 7 to 14. In contrast to the well-established markers cleaved-caspase-3 and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), many of the following markers are new and their validation and quality have been variably established. In addition, an alternative approach to defining the cell death mechanism is based on the application of chemicals and pharmacologic agents to cultured cells to stimulate or suppress certain mediators or pathways. The choice of adequate biomarkers intrinsically depends on the biological questions of each experiment or clinical investigation, with some of these markers being more reliable by providing consistent and reproducible results. While few markers that are specific to unique signaling pathways of RCD exist, most of the tests are useful if applied within standardized panels. Furthermore, it is important to mention that markers may be species-specific, thus hampering the ability to translate the results between different species.
Biochemical Features, Pathway Regulators, and Staining Markers for Different Types of Regulated Cell Death.
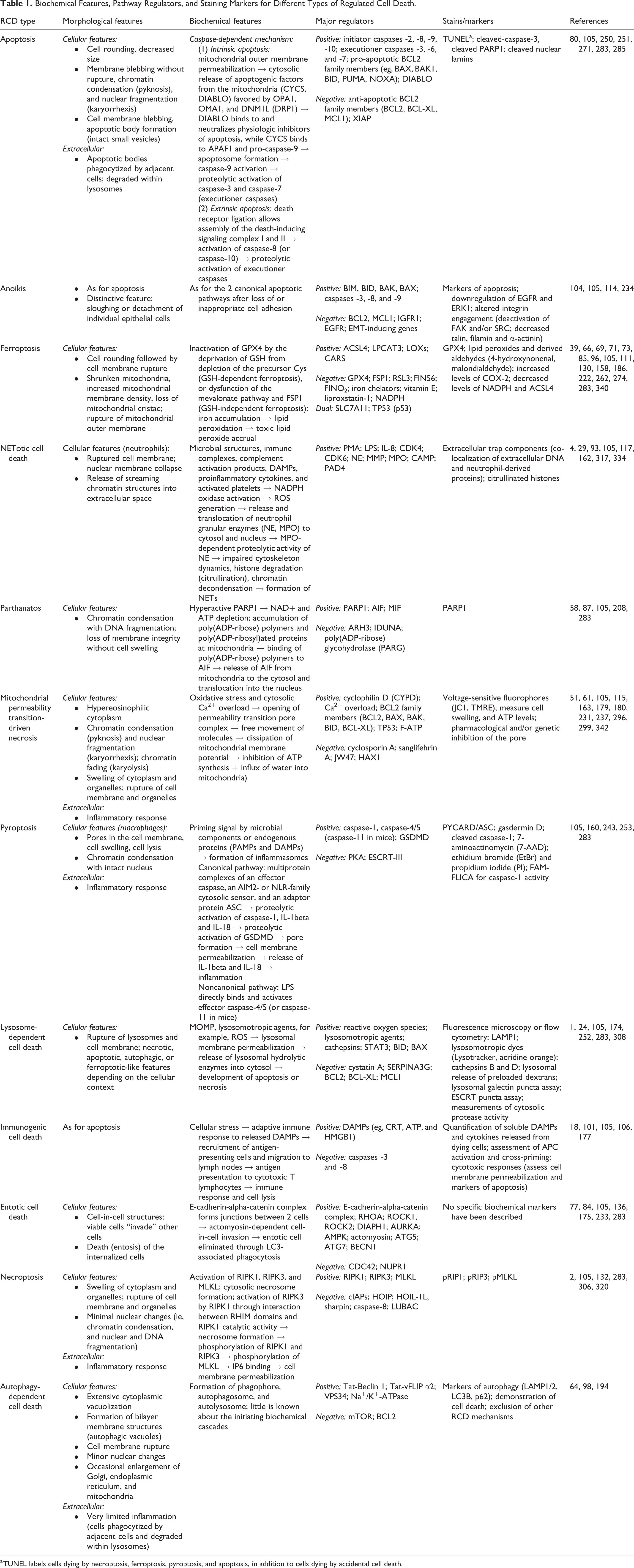
a TUNEL labels cells dying by necroptosis, ferroptosis, pyroptosis, and apoptosis, in addition to cells dying by accidental cell death.
Nuclear lamins are intermediate filament proteins that form the proteinaceous nuclear lamina surrounding the nucleus. Since the first event that leads to nuclear fragmentation involves the caspase-mediated proteolysis of lamins A, B, and C, these nuclear lamins can be used as a marker of apoptosis. 285
Conventional flow cytometry provides information about cell size and morphology to differentiate between viable and nonviable cells. Markers such as annexin V, propidium iodine (PI) or 4′,6-diamidino-2-phenylindole (DAPI) can be used for this purpose in conjunction with fluorescently tagged antibodies against target molecules implicated in RCD to provide insights into specific RCD mechanisms. 14
Due to its definition as apoptotic cell death induced by lack of correct cell/ECM attachment, biomarkers of anoikis mirror well-known markers of the intrinsic and extrinsic apoptotic pathways. Since the induction of anoikis requires lysosomal-mediated downregulation of EGFRs leading to termination of pro-survival signaling, 245 assessing EGFR level as well as inhibition of ERK1 can also help highlight the loss of cell anchorage. 104 In addition, the loss of cell anchorage due to altered integrin engagement can be assessed microscopically using fluorometric or colorimetric detection methods on cell culture plates using commercially available assays.
Quantification of the byproducts of lipid peroxidation (eg, 4-hydroxynonenal, malondialdehyde) is the most widely recognized indirect indicator of ferroptosis. 66 Identification of ACSL4 has been proposed as a reliable biomarker of ferroptosis given the gatekeeper role that this molecule plays in PUFA bioactivation. 340 Imbalances affecting important regulators of iron metabolism are also considered to be indicators of ferroptosis sensitivity. Low levels of NADPH imply sensitivity to ferroptosis across many cancer cell lines. 274 On the other hand, high expression of FSP1 correlates with ferroptosis resistance. 17 Cyclooxygenase-2 (COX-2) has also been associated with ferroptosis in the context of brain injury. 190
NETosis can be investigated using transmission electron microscopy, scanning electron microscopy, and immunofluorescence. On electron microscopy, NETs consist of a multitude of large interwoven threads up to 50-nm-diameter that branch into characteristic thinner threads of 15 to 17 nm diameter and globular domains of approximately 25 nm. 29 Co-localization of extracellular DNA and neutrophil-derived proteins, including MPO and NE, can be assessed in diseased tissues microscopically. Immunofluorescence as well as flow cytometric assays can be employed to determine NETosis by the detection of MPO and citrullinated histones in association with extracellular chromatin. High levels of cell-free DNA in bodily fluids are also a reliable indication of NETosis if accompanied by concurrent increase in DNA-histone complexes and neutrophil-derived proteins. 117
The identification of Parthanatos can be based on detecting the nuclear overexpression of PARP1 associated with nuclear translocation of AIF, large scale DNA fragmentation (∼50 kb, as opposed to small scale DNA fragmentation and laddering pattern typically produced by apoptosis), and absence of activation of execution caspases. 278
Most of what is known about markers of MPT-driven necrosis comes from research on myocardial cell death. 214 Several methods such as spectrophotometric measurement of Ca2+ or oxidant-induced swelling in mitochondria and Ca2+ retention capacity (CRC) assay have been established as indices of MPT pore opening in both isolated mitochondria and intact cells. 12 In addition, the loss of mitochondrial membrane potential can be assessed using voltage-sensitive dyes such as JC-1 or tetramethylrhodamine ethyl ester (TMRE); 61 or methods such as the calcein/cobalt quenching technique. 61,237 Given the inhibition of ATP synthesis, measurement of ATP levels can also be used as a surrogate. 214 No reliable in vivo markers exist to date.
The canonical pyroptosis pathway can be detected using immunohistochemistry for the ASC complex (apoptosis-associated speck-like protein containing a C-terminal caspase-recruitment domain [CARD]; often referred to as PYCARD), which is a major constituent of the inflammasome that relocates to the cytoplasm during pyroptosis. The downstream target GSDMD can detected by western blot using anti-GSDMD antibody. 160,243 Since both the caspase-1-dependent (ie, canonical) and caspase-1-independent (ie, noncanonical) pyroptotic pathways ultimately lead to the release of IL-1beta and IL-18, cytokine profiling can also aid to identify pyroptosis. In addition, the commercially available FAM FLICA assay can be used on frozen tissue sections or cells to assess caspase-1 activation. By electron microscopy, large oligomeric ring-shaped structures formed by the N-terminal GSDMD fragments can be visualized; the inner ring diameters of the GSDMD pores are estimated between 10 and 20 nm. 253
In LDCD, lysosomal membrane permeabilization can be assessed via fluorescence microscopy or flow cytometry. 24,308 Translocation of soluble lysosomal components (such as cathepsins) to the cytosol can be identified by detecting their location or by measuring cytosolic protease activity. Fluorescent dextrans and lysosomotropic agents (Lysotracker or acridine orange) can aid in visualizing lysosomal destabilization. Investigation of galectin translocation and ESCRT protein recruitment to damaged lysosomes are two other sensitive and novel methods. 308
In vivo indicators of potential ICD include cell surface CRT, the secretion of large amounts of ATP, and release of HMGB1 coinciding with cell death. 177 The gold standard for demonstration of ICD involves vaccination of mice with syngeneic tumor cells exposed to an ICD stimulus. Later transplant of live tumor cells in these mice should show deficient tumor growth and the effect should be absent in immunodeficient mice. 177
Although no specific markers have been discovered for entotic cell death to date, the location of the internalized cell within a lysosome can be identified by labeling with lysosomal-associated membrane protein (LAMP) 1, or fluorescent dyes for labeling and tracking acidic organelles. 206 The characteristic cell-in-cell phenomenon also has a concentric pattern of labeling with membrane markers such as E-cadherin in the engulfing and internalized cells. 131
Necroptosis is often detected with a combination of methods. 120 The formation of amyloid structures within the necrosome complex can be investigated using fluorochrome dyes such as thioflavin S and T, or Congo Red, or electron microscopy. 188 Transmission electron microscopy (TEM) may help identify the necrotic morphology characterized by swelling and vacuolization of the cytoplasm, swelling of the mitochondria, and rupture of the cell membrane. 43 Furthermore, Western blot or immunolabeling can be used to detect activation of RIPK1, RIPK3, and MLKL (ie, phosphorylation), formation of the necrosome, and MLKL oligomerization and translocation to the cell membrane identified using. 132,320
The morphologic hallmark of ADCD is the accumulation of double-membraned autophagosomes; however, this is not specific. 264 Various strategies to measure the flux of autophagy-associated proteins can be used. Autophagosomal markers such as LC3B and p62 and autolysosomal markers such as LAMP 1/2 are used to define autophagosome formation and maturation; when combined with the presence of cell demise, these only indicate ADCD if other RCD mechanisms can be excluded. 64
Conclusions and Remarks
The term RCD encompasses numerous processes in physiology and in pathologic conditions of animals and humans. Targeting specific RCD signaling pathways for the treatment of such conditions is of great interest to the scientific community. Although morphologic characterization of such entities was previously the only available option, the current availability of advanced biomolecular techniques allows a more reliable characterization and subclassification of RCD according to specific molecular signatures. Therefore, accurate identification of the type of cell death and correct use of RCD-related terms is advisable when supported by molecular evidence. Otherwise, if functional or biochemical characterization has not been done, the morphologic characteristics of dying cells do not accurately distinguish between different forms of RCD. It is therefore highly recommended to adopt a neutral term, such as “cell death” or “individual cell death,” to avoid confusion and misinterpretation when reporting histopathological findings.
While there have been some insights about RCD mechanisms in diseases of domestic animals based on in vivo or in vitro studies, current knowledge remains limited. As the RCD field advances and further assays become available, standard panel of biomarkers and functional tests should be used to characterize and describe pathways of interest and cell death mechanisms. Ultimately, the involvement of veterinary pathologists in studies on RCD is pivotal to accurately interpret cellular signals and differentiate cell death mechanisms by utilizing a standardized and comprehensive methodology that goes beyond the sole examination HE-stained tissue sections.
Supplemental Material
Supplemental Material, sj-pdf-1-vet-10.1177_03009858211005537 - Mechanisms of Regulated Cell Death: Current Perspectives
Supplemental Material, sj-pdf-1-vet-10.1177_03009858211005537 for Mechanisms of Regulated Cell Death: Current Perspectives by Sara Francesca Santagostino, Charles-Antoine Assenmacher, James C. Tarrant, Adeyemi O. Adedeji and Enrico Radaelli in Veterinary Pathology
Footnotes
Acknowledgements
The authors wish to acknowledge Dr Joshua D. Webster (Genentech, Inc) and Dr Lucia Minoli (University of Pennsylvania, Department of Pathobiology) for critically reviewing the manuscript and providing insightful comments and suggestions. All diagrams and diagrams included in this work have been composed using the Motifolio Toolkit for scientific illustrations (![]() ).
).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.