
Abstract
Coxiella burnetii, a highly adapted obligate intracellular bacterial pathogen and the cause of the zoonosis Q fever, is a reemerging public health threat. C. burnetii employs a Type IV secretion system (T4SS) to establish and maintain its intracellular niche and modulate host immune responses including the inhibition of apoptosis. Interactions between C. burnetii and caspase-1-mediated inflammasomes are not fully elucidated. This study confirms that C. burnetii does not activate caspase-1 during infection of mouse macrophages in vitro. C. burnetii–infected cells did not develop NLRP3 and ASC foci indicating its ability to avoid cytosolic detection. C. burnetii is unable to inhibit the pyroptosis and IL-1β secretion that is induced by potent inflammasome stimuli but rather enhances these caspase-1-mediated effects. We found that C. burnetii upregulates pro-IL-1β and robustly primes NLRP3 inflammasomes via TLR2 and MyD88 signaling. As for wildtype C. burnetii, T4SS-deficient mutants primed and potentiated NLRP3 inflammasomes. An in vivo model of pulmonary infection in C57BL/6 mice was developed. Mice deficient in NLRP3 or caspase-1 were like wildtype mice in the development and resolution of splenomegaly due to red pulp hyperplasia, and histologic lesions and macrophage kinetics, but had slightly higher pulmonary bacterial burdens at the greatest measured time point. Together these findings indicate that C. burnetii primes but avoids cytosolic detection by NLRP3 inflammasomes, which are not required for the clinical resistance of C57BL/6 mice. Determining mechanisms employed by C. burnetii to avoid cytosolic detection via NLRP3 inflammasomes will be beneficial to the development of preventative and interventional therapies for Q fever.
Coxiella burnetii, a zoonotic pathogen and cause of Q fever, is a well-adapted obligate intracellular bacterium that is highly infectious with a wide host range and global distribution. 56,78 Q fever manifests as epizootic abortions in naïve ruminant livestock herds, during which billions of infectious bacteria are deposited into the environment. 44,71 Resilient C. burnetii survive extracellularly for extended periods, travel by air to infect other hosts, including humans, via inhalation, and replicate within highly permissive alveolar macrophages. 19,34,88 Following inhalation of as few as 1 to 2 organisms, infected non-gravid ruminant hosts are asymptomatic yet intermittently shed bacteria. 44,71 Clinical outcomes in humans have been shown to be highly dependent on host immune responses as well as bacterial virulence, and range from asymptomatic to mild acute respiratory disease to potentially fatal endocarditis in at-risk patients. 41,51,69 Patients with acute Q fever may develop transient pneumonitis and splenic inflammation whereas those with abnormal immune responses develop a chronic form of the disease and may suffer from fulminant pneumonia, hepatitis, splenitis, and endocarditis. 41,56,69
Guinea pigs have been the predominant animal model to study Q fever because they develop a febrile response with clinical signs useful for evaluation of vaccine and therapeutic effects in vivo. 45,72 Mouse models have proven invaluable for elucidating immune responses to C. burnetii. Inbred mouse strains, particularly C57BL/6 mice, have an inherent resistance to C. burnetii due to effective bacterial killing both in vitro and in vivo. 13,79,88,90 Mouse bone marrow–derived macrophages (BMDM) are able to limit intracellular replication and development of the Coxiella-containing parasitophorous vacuole. 88,90 Notably, mouse alveolar macrophages are more permissive of C. burnetii similar to human and primate alveolar macrophages. 19,30 Recent studies have demonstrated that culture conditions of murine BMDM affect permissiveness to C. burnetii intracellular growth. 25 By utilizing wildtype inbred mouse strains and genetically engineered knockout counterparts, the roles of mammalian immune mechanisms and signaling pathways in response to C. burnetii infection are now better understood. 13,36,65,68,79,88,89
As for host susceptibility and resistance, virulence of C. burnetii strains also varies and is further complicated by phase variation. Phase variation is an evasion mechanism of Gram-negative bacteria, and in C. burnetii Nine Mile (NM) strain it results in a permanent chromosomal deletion within the encoding region of LPS. 7,39,40,73 This renders Nine Mile Phase II (NMII) strain avirulent (based on normothermia and lack of clinical disease in guinea pigs and immunocompetent mice), and exempt from Select Agent status. 60,72,80 Due to this exemption and because in vitro kinetics and cytokines responses to Nine Mile Phase I (NMI) and NMII strains are similar in mouse and human cells, NMII is used extensively in the study of host immune responses. 13,40 Nevertheless, the ultimate goal is to compare biochemical and physiologic responses in animal models infected with avirulent NMII to those infected the virulent C. burnetii strains.
Vital to the success of C. burnetii as an intracellular pathogen is its ability to avoid detection and modulate host cell immune responses, including inflammatory cytokine signaling and programmed cell death. 6,7,47,80,83 C. burnetii is exquisitely adapted to intracellular life, thriving within target cells of the monocyte-macrophage system. 80,83 Once C. burnetii are phagocytosed by macrophages, they actively commandeer host vesicular pathways for the development of a spacious, acidified, parasitophorous vacuole. 19,34 Host modulation by C. burnetii is dependent on secretion of bacterial (effector) proteins into host cytosol through a type IV secretion system (T4SS), common to other intracellular bacteria. 20,80,82 The T4SS is critical to parasitophorous vacuole formation and maintenance, intracellular replication, and inhibition of host cell death, all functions necessary to establish an intracellular niche. 6,62,63 Although over 130 different bacterial effector proteins are transferred from the parasitophorous vacuole into the host cytosol, only a few have known functions. 63,80 At least 2 T4SS proteins modulate programmed cell death pathways of the host cell. 28,52,53 AnkG, a eukaryotic-like, ankyrin repeat-containing protein, binds mitochondrial p32 leading to the inhibition of intrinsic apoptosis. 52,53 The more recently described inhibitor of caspase activation (IcaA) attenuates caspase-11-mediated pyroptosis, reducing pore formation, and inhibiting NLRP3 inflammasomes and caspase-1 activation. 28,52,80 Recent reports also describe direct inhibition of cytokine signaling and autophagy pathways by C. burnetii. 24,48
In contrast to apoptosis, a noninflammatory form of caspase-mediated programmed cell death, pyroptosis is pro-inflammatory and mediated by the effector cysteine protease caspase-1, which is central to multiprotein complexes known as inflammasomes. 10,31,35 Formation of canonical inflammasomes within the cytosol leads to recruitment and cleavage of pro-caspase-1 to its active form. Caspase-1 activation leads to secretion of the pro-inflammatory cytokines, IL-1β and IL-18 and pyroptosis. 15 Inflammasome formation is the result of cytosolic sensing of myriad stimuli by intracellular receptors, such as NLRP1/NALP1b, NLRC4/IPAF, NLRP3/NALP3, and AIM2. 35 Some of these receptors have numerous ligands, while others have only few known stimuli, for example, NLRC4/IPAF detects bacterial flagellin and components of Type III secretion systems. 16,35
The NLRP3 inflammasome is a well-characterized canonical inflammasome and is activated by several microbial products including bacterial toxins and viral RNAs, as well as nonmicrobial substances such as uric acid, reactive oxygen species, ionic imbalances, and extracellular matrix components. 3,5,14,15,33,35 NLRP3 inflammasome activation requires 2 signals: the first (signal 1) is referred to as “priming,” and involves Toll-like receptor (TLR) signaling and NFκB-mediated upregulation of NLRP3 and pro-forms of IL-1β and IL-18. 42,66 Signal 2 involves sensing of aforementioned stimuli, resulting in aggregation of NLRP3 proteins, and co-localization with ASC (apoptosis-associated speck-like protein containing a carboxy-terminal caspase activation and recruitment domain [CARD]), a bipartite adaptor protein that bridges NLRP3 and pro-caspase-1. 14,43,61 Once pro-caspase-1 is recruited to the NLRP3-ASC focus, it is cleaved and activated. Active caspase-1 subsequently cleaves pro-forms of IL-1β and IL-18 and induces pore formation via the inflammasome protein gasdermin D, resulting in pyroptosis. 15,35,49
Inflammasomes are considered critical to the development of an inflammatory response sufficient to combat microbial infections and for the clearance of intracellular pathogens. 15,26,58,76 It is not surprising that successful intracellular pathogens have evolved mechanisms to subvert inflammasome activation through avoidance of detection or direct inhibition of inflammasome components. 29,46,54 Examples include the downregulation of flagellin by Salmonella enterica serovar Typhimurium to avoid detection by NLRC4, and the T3SS effector YopM of Yersinia pseudotuberculosis, which binds to pro-caspase-1 and inhibits its recruitment to the forming inflammasome. 29,32,46,54 Recent studies have demonstrated that C. burnetii does not induce caspase-1 activation in mouse macrophages. 13,28 It was also found that C. burnetii possess a T4SS effector, IcaA, capable of attenuating caspase-1 activation through the noncanonical caspase-11-mediated pathway. 28 However, the influences of C. burnetii and canonical inflammasomes on each other as well as the relevance of inflammasomes during natural infection remain to be elucidated.
In this study, the relationships between NMII C. burnetii and host inflammasomes were investigated using an in vitro model developed with BMDM from C57BL/6 mice and an in vivo model using wildtype and mice-deficient in caspase-1 and NLRP3.
Materials and Methods
Cell Culture
Primary bone marrow–derived macrophages (BMDM) were obtained from wildtype (WT) C57BL/6J mice (Jackson Laboratories) or mice with the following knockout mutations (MGI reference ID included): Tlr2tm1Aki (Tlr2−/−) MGI:2178675, Tlr4tm1Aki (Tlr4−/−) MGI:1860885, combined Tlr2tm1Ak and Tlr4tm1Aki (Tlr2/4−/−), Myd88tm1Aki (MyD88−/−) MGI:2385681, Ticam1tm1Aki (Trif−/−) MGI:3029019, Casp1tm1Flv (Casp1−/−) MGI:2158744, Nlrp3tm1Vmd (Nlrp3−/−) MGI:3622094, Nlrc4tm1Vmd (Nlrc4−/−) MGI:3046934. BMDM were cultured in DMEM (Dulbecco’s modified Eagle medium; Invitrogen) supplemented with 10% fetal calf serum (FCS), 5 mM HEPES, 0.2 mg/ml L-glutamine, 0.05 mM 2-ME, 50 µg/ml gentamicin sulfate, and 10 000 U/ml penicillin and streptomycin with 30% L cell-conditioned medium. Knockout mice were gifted from the laboratories of Dr E. Miao of University of North Carolina at Chapel Hill (Nlrp3−/− and Nlrc4−/−), Dr C. Roy of Yale University (Casp1−/−), and Drs W. Altemeier, M. Gale, and A. Haijar of University of Washington (Tlr2−/−, Tlr4−/−, and Tlr2/4−/−, MyD88−/−, and Trif−/−). 1,21,59,86,87 All mice were housed and maintained at University of Washington and backcrossed at least 6 generations onto a C57BL/6J background. Propagated macrophages were harvested using cold PBS (phosphate-buffered saline) containing 1 mM EDTA (ethylenediaminetetraacetic acid), suspended in phenol red-free antibiotic free DMEM supplemented with 5% or 10% FCS depending on length of infection or treatment. When indicated, LPS stimulation was achieved by treating macrophages with Salmonella minnesota R595 lipopolysaccharide (LPS; 100 ng/ml; List Biologicals) for either 4 hours or approximately 18 hours prior to infections or treatment with inflammasome stimuli. Experiments measuring IL-1β release in the supernatants, caspase-1 activation or foci formation were performed in the presence of 5 mM glycine to reduce cell lysis. 31
Bacteria
All in vitro Coxiella infections were achieved using NMII (RSA clone 439) C. burnetii (wildtype, mCherry-expressing, or T4SS-deficient [▵dotA]) at multiplicities of infection (MOI) of 20 unless otherwise indicated. Bacteria were diluted in cell media based on genome equivalents of frozen stocks, added to plate wells, then centrifuged onto cells for 3 minutes at 150 × g. Salmonella enterica serovar Typhimurium (Stm; SL1344) infections were performed at MOI 10 for 1 hour following growth in LB overnight at 37 °C, dilution of 1:15, and growth for 3 hours in LB containing 0.3 M NaCl. Yersinia pseudotuberculosis (Yptb; JOEHKM- 46 ) infections were performed at MOI 20 for 90 minutes following growth overnight at 25 °C in LB, dilution of 1:40 into LB with 20 mM MgCl2 and 20 mM Na2C2O4, and growth at 25 °C for 1 hour and then 37 °C for 2 hours. Both Salmonella and Yersinia were then washed in PBS, quantified using a Beckman Coulter Multisizer 4, then added at appropriate MOI, and spun onto macrophages for 3 minutes at 150 × g.
Reagents and Assays
To induce NAIP5/NLRC4 inflammasomes, LPS-stimulated cells were infected with Stm. For NLRP3-specific caspase-1 activation, 10 μM nigericin (a purified bacterial toxin; Sigma-Aldrich) and Yptb (JOEHKM-) were used following 4 or 18 hours of LPS stimulation, respectively. Cytotoxicity was assessed by measuring lactate dehydrogenase (LDH) release into cell supernatants (CytoTox 96 Non-Radioactive Cytotoxicity Assay, Promega). IL-1β-secretion was measured in cell supernatants using a sandwich ELISA assay (DuoSet, R&D Systems) following filtration by 0.22-mm filter (Corning).
Immunocytochemistry, Caspase-1 Detection, and Microscopy
To evaluate inflammasome foci in vitro, mouse macrophages were seeded onto 12 mm coverslips within 24-well plates (Corning). Following infections and treatments, cells were washed with 0.1% bovine serum albumin (BSA) in PBS and then fixed with 4% paraformaldehyde or washed, fixed, and permeabilized using the Cytofix/Cytoperm kit (BD Biosciences). Primary antibodies used were NLRP3 (Santa Cruz; sc-66846) and ASC (Millipore; 04-147) with species-targeted fluorescent secondary antibodies (AlexaFluor). To evaluate the presence of active caspase-1 in cells, the FAM-YVAD-FMK caspase-1 detection assay (FLICA, ImmunoChemistry Technologies, LLC) was used according to manufacturer’s instructions. Nuclear visualization was achieved through Hoechst staining. All cells were similarly washed; coverslips mounted on slides, sealed, and then visualized using a Leica SL Confocal microscope or DeltaVision Elite Microscopy Imaging System (GE Healthcare). At least 10 separate high power (400×) fields were evaluated to determine presence of NLRP3, ASC, or active caspase-1 (FLICA) foci. Images of representative fields were captured at 400× or 600× magnification.
Immunoblot Analysis
Macrophages were lysed with 1× SDS sample buffer (Invitrogen) and then eluted with boiling at 100 °C for 10 minutes. Lysates were separated by SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membrane, and detected by the indicated antibodies, Immobilon chemiluminescence system (Millipore), and Odyssey (Li-Cor). Primary antibodies used included NLRP3 (Cell Signaling; D4D8T), NLRC4 (Millipore; 06-1125), pro-IL-1β (R&D Systems; AF-401-NA), and actin (Abcam; ab6276). Target protein relative fluorescent units (RFU) were normalized to actin.
Mice and Pulmonary Infections
Wildtype (WT) C57BL/6J and knockout (KO; Casp1−/− and Nlrp3−/−) mice as described above (Cell Culture) were bred and housed at University of Washington (UW) School of Medicine, an American Association for the Accreditation of Laboratory Animal Care (AAALAC) accredited facility. All experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC). Mice were co-housed with 2 to 5 mice per cage in standard plastic cages with corn-cob bedding and nestlet material provided for enrichment. Following infection with C. burnetii, mice were housed under biosafety level-2 (BSL-2) conditions.
Twelve- to 16-week-old male and female mice (n = 4–8 per genotype per time point) were infected with NMII (RSA clone 439) Coxiella burnetii (1 × 107 bacteria) suspended in 50 µl PBS via oropharyngeal instillation under isoflurane anesthesia. Control mice (n = 3–6 per genotype) were instilled with PBS only. Mice were monitored for clinical signs such as lethargy, hunched posture, ruffled fur, or increased respiratory rate and effort, throughout infection. Mice were weighed at 3, 7, 14, and 21 days postinfection and those at endpoint were sacrificed via isoflurane anesthesia and exsanguination via cardiocentesis. The left bronchus was tied off using monofilament suture, and the left lung removed and frozen at −80 °C. The remaining right lung was instilled at 20 mm Hg pressure and immersion fixed in 10% neutral buffered formalin (NBF). Spleens were weighed and compared to body weight; a portion was immersion fixed in 10% NBF.
Molecular Quantification of Pulmonary Bacterial Burdens
Coxiella-infected and PBS-instilled (control) mouse lungs were snap frozen in liquid nitrogen and stored at −80 °C until processed. Purification of total lung DNA was completed using Qiagen’s DNeasy Blood & Tissue Kit according to manufacturer’s instructions that included addition of an RNase digestion step. Briefly, 25 ng of frozen lung tissue was homogenized using ceramic beads in an Omni bead homogenizer; the DNA extracted and purified, quantified using a NanoDrop spectrophotometer, and diluted in nuclease-free water to a final concentration of 100 ng/μl. Absolute quantitative PCR was performed on 300 ng purified genomic DNA in quadruplicate using a 384 well format ABI 7900 HT platform. Primers and probe targeting C. burnetii dotA gene were used (F: GCGCAATACGCTCAATCACA, R: CCATGGCCCCAATTCTCTT, and probe: CCGGAGATACCGGCGGTGGG; IDT), as previously described. 27 TaqMan Universal PCR Master Mix was used for amplification using ABI universal cycling conditions. C. burnetii copy number was determined as previously described 27,88 using full-length C. burnetii dotA sequence inserted into pCR 2.1-TOPO plasmid (Invitrogen TOPO TA Cloning kit). Purified plasmid was quantified by NanoDrop spectrophotometer and diluted in nuclease-free water to give 108 copies/μl. Log10 dilutions were made in quadruplicate to yield 108 to 100 copies/reaction. R2 values for log10 copy number versus Ct in the standard curves were >0.99. Uninfected control lungs routinely exhibited no amplification through 40 PCR (polymerase chain reaction) cycles in this assay. Copy number data are expressed as C. burnetii genome copies/µg isolated lung DNA.
Histology, Immunohistochemical Staining, and Quantitative Microscopy
Following immersion fixation in 10% neutral buffered formalin, lung, liver, and splenic tissues were routinely processed, sectioned, and stained for histologic (hematoxylin and eosin) and immunohistochemical analyses. Histologic changes were evaluated by a board-certified veterinary pathologist (M. A. Delaney). Polyclonal antibody to C. burnetii NMII (gift from Robert Heinzen) and F4/80 clone BM8 (Invitrogen, MFF48000) were applied to lung tissue sections at 1:1000 and 1:200, respectively, and detected using 3,3′-diaminobenzidine (DAB) chromogen in a Leica Bond-Max Automated Immunostainer. Following counterstaining with hematoxylin and coverslipping, the slides were scanned in brightfield at 200× magnification using the Hamamatsu NanoZoomer Digital Pathology System. The digital images were then imported into Visiopharm® software for quantitative analysis. Using the Visiopharm® Image Analysis module, regions of interest (ROIs) were manually drawn around lung and spleen in each sample to include 100% of the respective tissue section. The software converted the initial digital image into grayscale values using 2 features, RGB-B and HDAB-DAB. Visiopharm® software was then trained to label positive C. burnetii (NMII) or F4/80 staining and the hematoxylin counterstain using a threshold of pixel values. All ROIs were processed in batch mode using this configuration to generate the desired outputs as a ratio of positive staining over total area (ROI) evaluated.
Statistical Analysis
GraphPad Prism software was used to calculate statistical significance by 1- or 2-way ANOVA (*P < .05, **P < .01, ***P < .001) with Bonferonni (multiple comparisons) posttests, unless otherwise indicated. Data from in vitro experiments are representative of at least 3 independent experiments.
Results
NMII C. burnetii Develops a Permissive Infection in C57BL/6 BMDM by 24 Hours In Vitro
To study interactions between C. burnetii and host inflammasomes an in vitro model of the early phase of infection was established. This model verified NMII C. burnetii can infect BMDM from wildtype C57BL/6J mice. BMDM were infected with mCherry-expressing NMII at multiplicity of infection (MOI) 20. After 24, 48, and 72 hours of infection, NMII were detected within BMDM by microscopy (Fig. S1b, c, d). Uninfected cells did not contain bacteria (Fig. S1a). Infection for 24 hours was identified as an appropriate time point for further studies as most BMDM contained bacteria at that time (Fig. S1b). Following 72 hours of infection, a large percentage of macrophages contained aggregates of viable intracellular bacteria indicative of persistent infection (Fig. S1d).
NMII C. burnetii Does Not Induce Caspase-1 Activation, IL-1β Secretion, or Pyroptosis In Vitro
To determine if C. burnetii activates caspase-1 in the in vitro model, BMDM from WT C57BL/6J mice were infected with NMII at MOI 20. Following 24 hours of infection, caspase-1 activation was determined by the presence of green fluorescent foci using FAM-YVAD-FMK caspase-1 detection assay (Fig. 1a–e). For a positive control in all studies, robust caspase-1 activation was achieved by treating the cells with nigericin (10 µM) for 30 minutes following 4 hours of LPS-priming to induce NLRP3 inflammasome activation (Fig. 1c) or infecting LPS-stimulated (18 hours) BMDM with Salmonella enterica serovar Typhimurium (Stm) at MOI 10 for 1 hour to stimulate the NLRC4 inflammasome (Fig. 1d). For a negative control, LPS-stimulated BMDM were left untreated/uninfected (Fig. 1a). In contrast to Stm-infected and nigericin-treated cells, in which a large percentage contained discrete green fluorescent cytoplasmic (active capase-1) foci accompanied by nuclear condensation (Fig. 1c, d; arrows), NMII-infected cells did not differ from uninfected cells with normal nuclear detail and no detectable foci (Fig. 1b). Caspase-1 activation was quantified as percentage of cells containing green fluorescent foci (Fig. 2a).
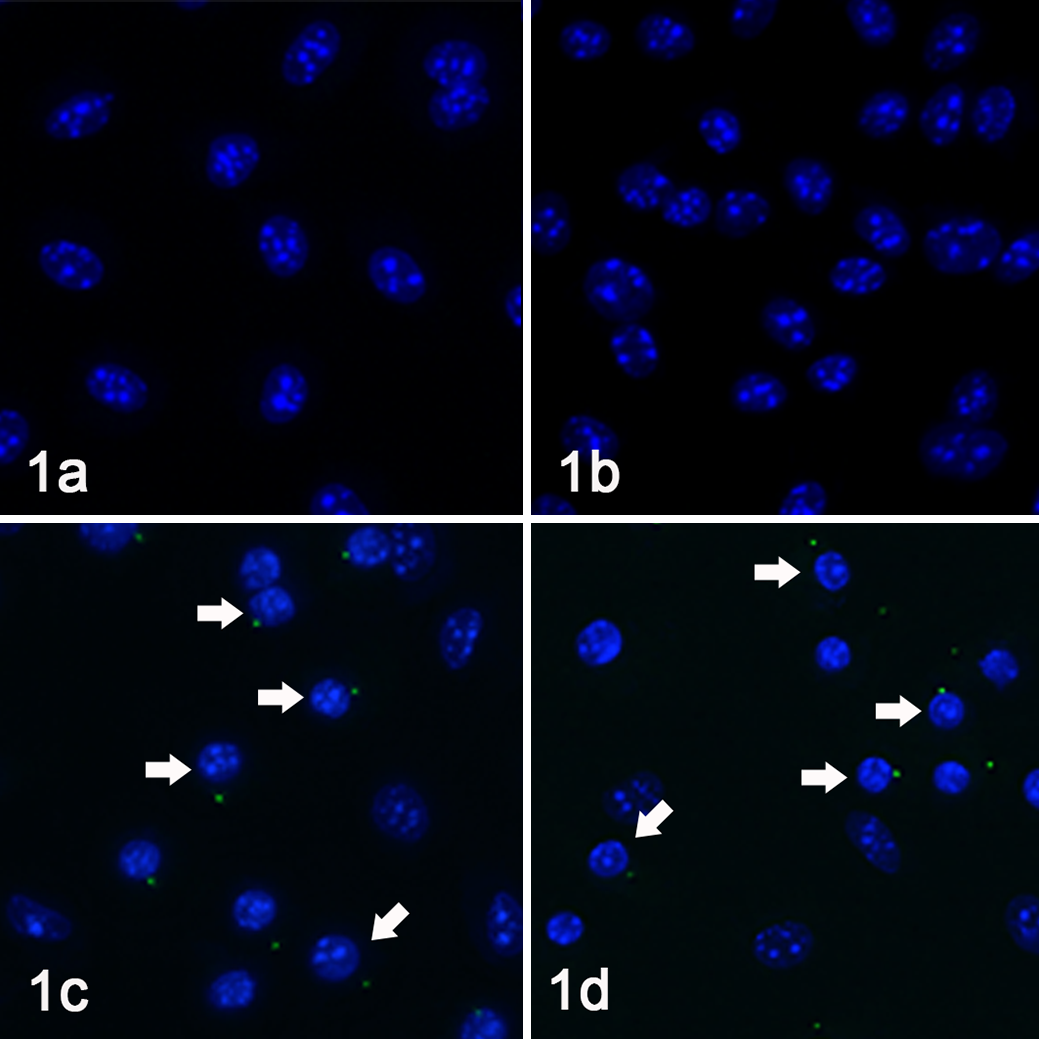
Macrophages, mouse, fluorescence microscopy for caspase-1. Caspase-1 activation (foci formation) was visualized using FAM-YVAD-FMK caspase-1 detection assay (green fluorescence). Nuclear morphology was monitored using Hoechst staining (blue fluorescence). Coxiella-infected (a) and uninfected (b) bone marrow–derived macrophages (BMDM) did not exhibit active caspase-1 foci within the cytoplasm. In contrast, many nigericin-treated (c) and Stm-infected (d) macrophages contained intense green cytoplasmic foci, often adjacent to a condensed (pyroptotic) nucleus (arrows).
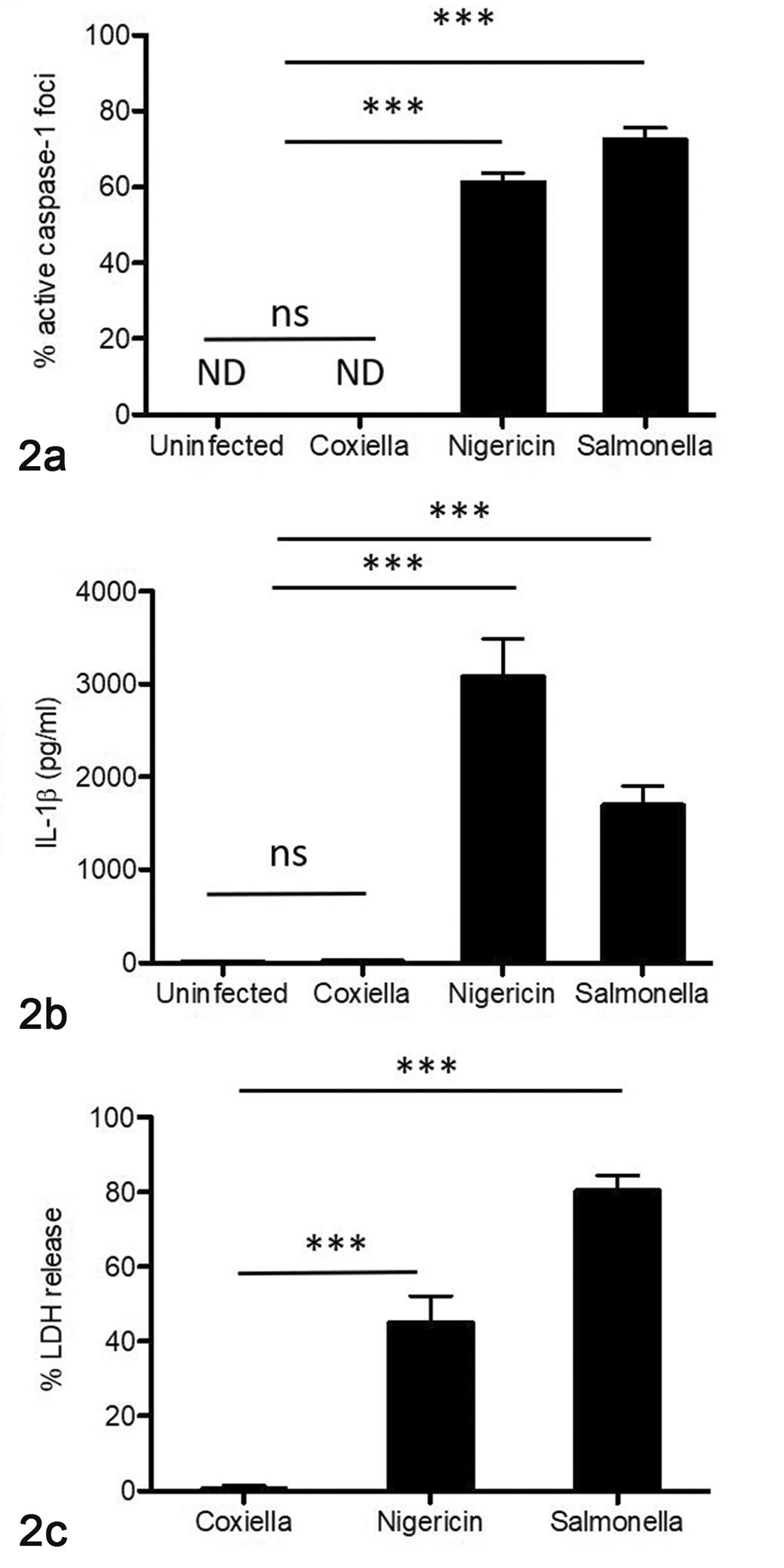
Caspase-1 foci quantification, IL-1β secretion, and cytotoxicity. (a) Percentages of uninfected, NMII Coxiella-infected, Stm-infected, and nigericin-treated bone marrow–derived macrophages (BMDM) containing active caspase-1 foci were quantified. (b) Uninfected and Coxiella-infected BMDM secreted negligible levels of IL-1β, in contrast to nigericin-treated and Stm-infected BMDM, which secreted large amounts of IL-1β. (c) Compared to spontaneous cell death of uninfected macrophages, nigericin-treated and Stm-infected BMDM had significantly increased cytotoxicity as measured by lactate dehydrogenase (LDH) release. Coxiella-infected BMDM had zero to minimal cytotoxicity. Data represents at least 3 individual experiments. The data show the mean ± standard error of the mean (SEM). One-way ANOVA with Bonferroni’s multiple comparison test, (**) P < .001, (***) P < .0001, ns = not significant, ND = not detected.
As confirmation of lack of caspase-1 activation by NMII C. burnetii IL-1β was measured via sandwich ELISA and cell death (ie, cytotoxicity) was measured by LDH release in supernatants from uninfected and NMII-infected BMDM. Nigericin-treated and Stm-infected BMDM were utilized as indicators of intact NLRP3 and NLRC4 inflammasome pathways, respectively. As expected, high concentrations of IL-1β and LDH were detected in supernatants of nigericin-treated and Stm-infected cells (Fig. 2b, c). In contrast, NMII-infected cells secreted negligible amounts of IL-1β and exhibited no cytotoxicity following 24 hours (Fig. 2b, c), which persisted throughout 72-hour infections (data not shown).
Together these results demonstrated that NMII C. burnetii infection in mouse BMDM did not result in caspase-1 activation, IL-1β secretion, or cell death during the first 72 hours of infection in vitro.
NMII C. burnetii Infection Does Not Induce NLRP3-ASC Foci Formation In Vitro
Since C. burnetii lacks a gene encoding flagellin (a known NLRC4 agonist), but has a T4SS that secretes bacterial proteins and products into the cytosol, we turned our focus to the NLRP3 inflammasome. 34 To determine if C. burnetii or its T4SS products are sensed within the host cytosol but can block activation of caspase-1, NLRP3-ASC foci formation was evaluated. BMDM from wildtype mice were infected with NMII (MOI 20, 24 hours) and then labeled with antibodies targeting NLRP3 (Fig. 2a, e, i) and ASC (Fig. 2b, f, j). In addition, FAM-YVAD-FMK (FLICA assay) was used to detect active caspase-1 (Fig. 2c, g, k). To demonstrate intact inflammasome pathways, BMDM were treated with nigericin (positive control; Fig. 2i–l) or left uninfected (negative control; Fig. 2a–d). Nuclei were visualized with Hoescht (Fig. 2d, h, l).
As expected, BMDM treated with nigericin had prominent ASC foci (Fig. 3j) that colocalized with NLRP3 (Fig. 3i) and active caspase-1 (Fig. 3k) confirming cytosolic detection (ie, signal 2), inflammasome formation, and caspase-1 activation (Fig. 3l). Cells with inflammasome foci (NLRP3-ASC-caspase-1) often had condensed nuclei representing pyroptosis (Fig. 3l). Like uninfected cells (Fig. 3a–d, upper panel), NMII-infected cells had no evident NLRP3 or ASC foci indicating a lack of cytosolic detection (Fig. 3e–h). Consistent with previous results, NMII-infected cells contained no active caspase-1 foci and had normal nuclei as for uninfected cells. These findings demonstrate that NMII C. burnetii did not induce NLRP3-ASC foci formation, signifying that neither these bacteria nor their products were detected by NLRP3 within the cytosol. This suggests that by avoiding cytosolic detection by NLRP3, there is no signal 2, thus no recruitment or cleavage of pro-caspase-1, no activation of caspase-1, and no subsequent pyroptosis or IL-1β secretion.
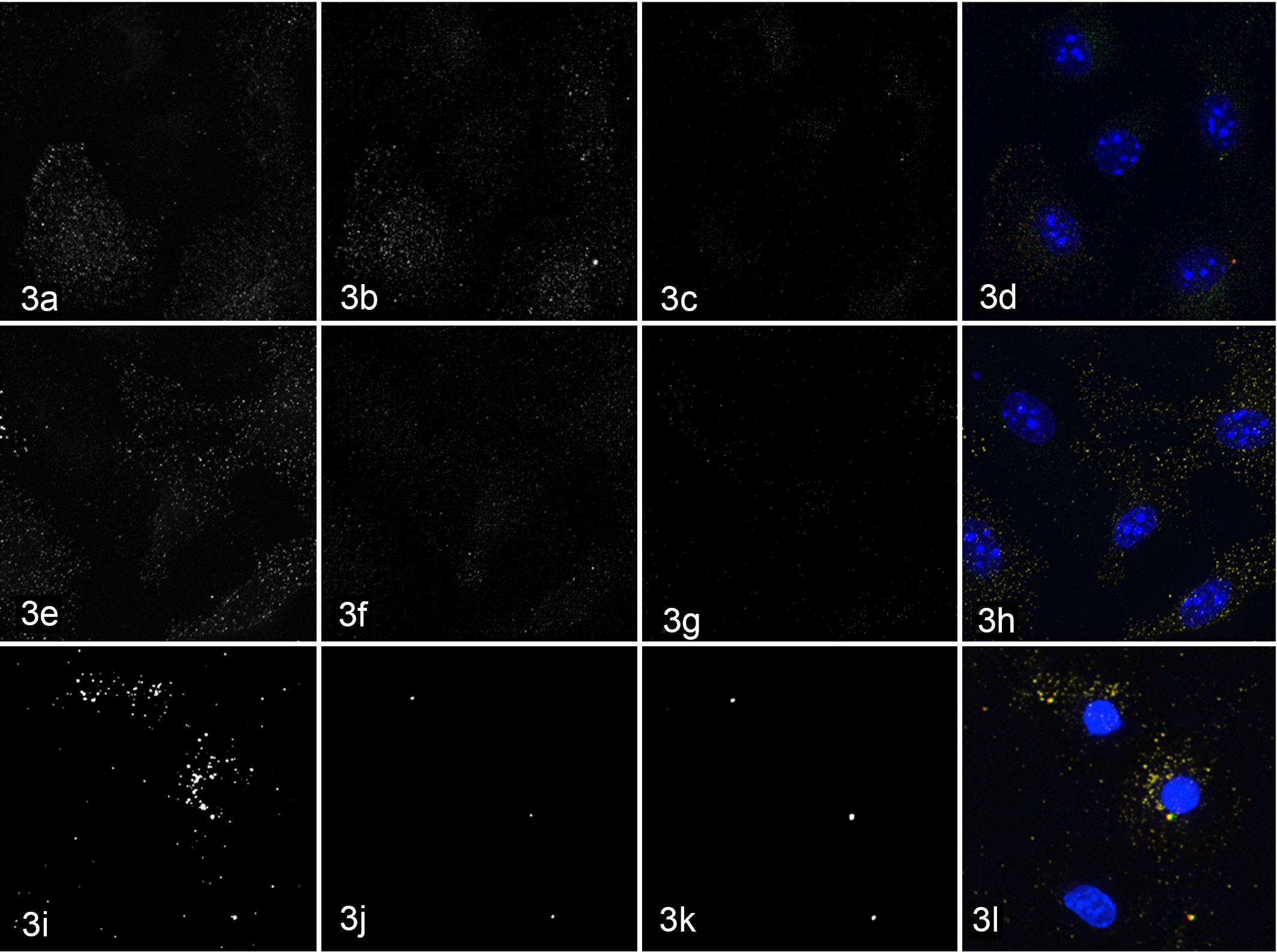
Macrophages, mouse. Immunofluorescence microscopy for NLRP3 inflammasome components. Uninfected (a–d), NMII Coxiella-infected (e–h), and nigericin-treated (i–l) bone marrow–derived macrophages (BMDM) were labeled with antibodies targeting the cytosolic sensor NLRP3 (a, e, i) and the adaptor protein ASC (b, f, j) in addition to staining with FAM-YVAD-FMK (FLICA, active caspase-1; c, j, k) and Hoechst (DNA, blue; d, h, l). Nigericin-treated macrophages have aggregates of NLRP3 with multifocal co-localization with both ASC (j) and active caspase-1 (k), with condensed (pyroptotic) nuclei (l), representing inflammasome formation. In contrast, uninfected and Coxiella-infected BMDM have no NLRP3 (a, e), ASC (b, f), or active caspase-1 (c, g) staining.
NMII C. burnetii Is Unable to Inhibit and Instead Potentiates Caspase-1-Mediated Cell Death and IL-1β Secretion in the Presence of Potent Inflammasome Agonists
Since NMII C. burnetii does not activate inflammasomes or caspase-1 in vitro, we wanted to test if NMII is able to inhibit caspase-1 activation in the presence of potent inflammasome stimuli. Yersinia pseudotuberculosis (Yptb) was included as another potent NLRP3 stimulus but is a live bacterium in contrast to nigericin, a purified bacterial toxin. Naive BMDM from WT mice were either pre-infected with NMII (MOI 20, 24 hours) or left uninfected, and subsequently infected with Stm to induce NLRC4/IPAF inflammasomes. LPS-stimulated BMDM were similarly pre-infected with NMII (or not), then either treated with nigericin or infected with Yptb to induce NLRP3 inflammasomes. For negative controls, naïve and LPS-stimulated BMDM were left uninfected or untreated. To confirm that Stm-, Yptb-, and nigericin-induced cytotoxicity and IL-1β secretion were caspase-1-mediated, BMDM from C57BL/6 mice deficient in caspase-1 (Casp1−/ − ) were utilized in parallel experiments.
As expected, BMDM infected with Stm and Yptb (JOEHKM-) and those treated with nigericin had large amounts of LDH released into supernatants (ie, cytotoxicity) and high levels of IL-1β secretion when compared to uninfected and NMII-infected BMDM (Fig. 4) Surprisingly, WT BMDM that were NMII-infected prior to Stm or Yptb infection or nigericin treatment had significantly higher cytotoxicity than those cells infected with only Stm or Yptb or treated with nigericin alone (Fig. 4a, c, e). As expected, all Casp1−/− BMDM had minimal or no measurable cytotoxicity (Fig. 4a, c, e). Similarly, uninfected BMDM and those infected with only NMII had negligible or undetectable levels of IL-1β in the supernatants (Fig. 4b, d, f). Pre-infection with NMII resulted in significantly higher IL-1β secretion from BMDM subsequently infected with Stm and Yptb or treated with nigericin (Fig. 4b, d, f). Casp1 − BMDM had minimal or no measurable IL-1β secretion (Fig. 4b, d, f).
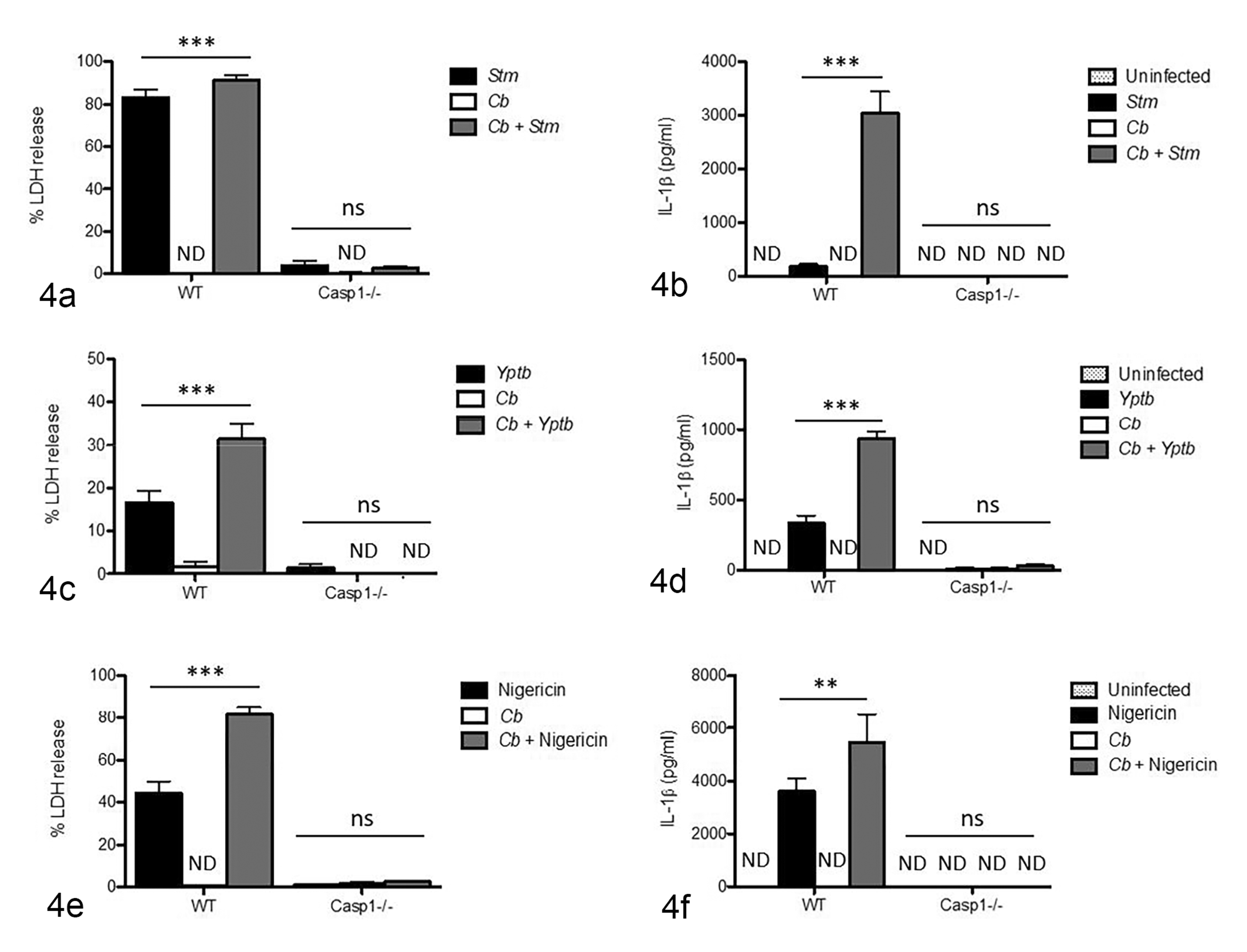
Cytotoxicity and IL-1β secretion. (a, b) Naïve bone marrow–derived macrophages (BMDM) were infected with Salmonella (Stm; black bars), infected with only NMII Coxiella burnetii (Cb; white bars), or pre-infected with NMII then infected with Stm (gray bars). (c, d) LPS-stimulated BMDM were infected with Yersinia (Yptb; black bars), infected with only NMII (white bars), or pre-infected with NMII and then infected with Yptb (gray bars). (e, f) LPS-stimulated BMDM were treated with nigericin (black bars), infected with only NMII (white bars), or pre-infected with NMII and then treated with nigericin (gray bars). (a, c, e) NMII-pre-infected BMDM had increased cytotoxicity compared to BMDM infected with Stm or Yptb or treated with nigericin alone, as measured by lactate dehydrogenase (LDH) release. No cytotoxicity is detected in NMII-infected BMDM. Minimal or no cytotoxicity is detected in Stm- or Yptb-infected or nigericin-treated BMDM from Casp1−/− mice. (b, d, f) IL-1β secretion is significantly elevated in Stm-, Yptb-infected, and nigericin-treated BMDM pre-infected with NMII, compared to BMDM only infected with Stm or Yptb or treated with nigericin. No IL-1β secretion is detected in uninfected and NMII-infected BMDM or in BMDM from Casp1 − mice. Data represent at least 3 individual experiments. Two-way ANOVA with Bonferroni’s multiple comparison test, (**) P < .001, (***) P < .0001, ns = not significant, ND = not detected.
Since Stm has been described to activate both NLRC4 and NLRP3, we wanted to verify which pathway was responsible for our results. 18,55,67 To address this, similar co-infection experiments with NMII using BMDM from mice deficient in NLRP3 or NLRC4 were performed. Results showed that the first hour of Stm infection NLRC4 was critical to pyroptosis and IL-1β processing while NLRP3 was not (Fig. S2a and b).
These data showed that cell death (ie, pyroptosis) and IL-1β secretion during Stm or Yptb infection and following nigericin-treatment were mediated by caspase-1. Furthermore, rather than attenuating inflammasome formation, pre-infection with NMII C. burnetii potentiated caspase-1-mediated (inflammasome) processes in vitro. These data also demonstrated that NMII was unable to inhibit activation of either the NLRC4 or NLRP3 inflammasomes (or subsequent caspase-1 activation) in the presence of canonical stimuli. Importantly, NMII potentiated caspase-1 activation induced by several different inflammasome agonists, thus it appears that NMII does not act directly upon caspase-1.
NMII C. burnetii Primes NLRP3 Inflammasomes
Since increased inflammasome activation in NMII-pre-infected cells was observed, we next wanted to determine if NMII C. burnetii can provide signal 1 (priming) in NLRP3 inflammasomes. To do this, levels of inflammasome components were measured in BMDM from WT mice infected with NMII or left uninfected via immunoblotting. During the last 4 hours of infection, BMDM were either LPS-treated or left untreated. Cell lysates were separated by SDS-PAGE and probed for NLRP3 and the pro-form of IL-1β. As previously shown, we observed that 4 hours of LPS treatment led to increased NLRP3 levels in uninfected cells. Following 24 hours of infection, NMII-infected BMDM lysates contained increased NLRP3 protein levels compared to uninfected cells with or without LPS treatment (Fig. 5a, b). Pro-IL-1β protein was not detected in naïve uninfected BMDM. With LPS treatment, there was an increase in pro-IL-1β in both uninfected and NMII-infected BMDM (Fig. 5a, c). A slight increase in pro-IL-1β following NMII infection alone was also evident (Fig. 5a, c). There appeared to be an additive effect with higher protein levels in BMDM infected with NMII and then treated with LPS (Fig. 5a, c). Since we saw increased NLRC4-dependent responses to Stm infection, we measured NLCR4 levels in uninfected and NMII-infected BMDM in the presence and absence of LPS. We found that NMII C. burnetii and LPS induced mild increases in NLRC4 compared to uninfected and untreated BMDM (Fig. 5d, e). These data demonstrated that NMII C. burnetii primed NLRP3 and NLRC4 inflammasomes during early infection with robust upregulation of NLRP3 and with lesser upregulation of pro-IL-1β in the absence of LPS.
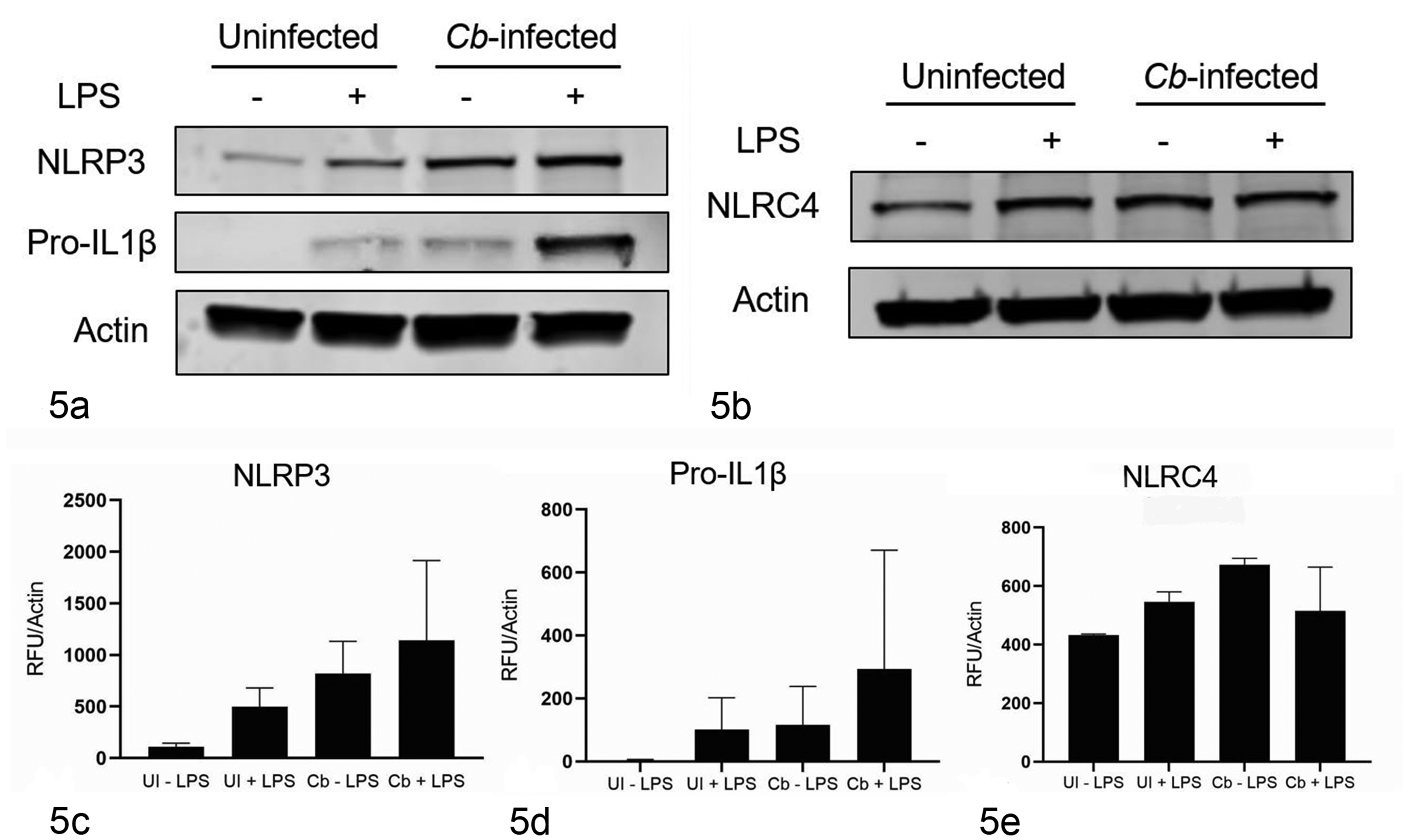
Immunoblot for inflammasome components. (a) NMII C. burnetii (Cb)-infected bone marrow–derived macrophages (BMDM) lysates contain increased NLRP3 and pro-IL-1β levels in the absence of LPS stimulation compared to uninfected, naïve BMDM and uninfected, LPS-stimulated (UI, +) BMDM. (c) Actin normalization highlights the increased NLRP3 levels seen in Cb-infected BMDM subsequently treated with LPS. (d) Cb-infected BMDM lysates also have slightly increased levels of pro-IL-1β compared to uninfected BMDM. However, LPS-treated BMDM have much higher pro-IL-1β levels comparatively. (b, e) In contrast, NLRC4 levels were similar among LPS-treated and NMII-infected BMDM but higher than uninfected, naïve BMDM. Graphs are representative of at least 2 (c, e) or 3 (b) individual experiments. The data show the mean ± standard error of the mean (SEM).
Upregulation of NLRP3 by NMII C. burnetii Is Dependent on TLR2-MYD88 and Independent of TLR4 and TRIF Signaling
To elucidate signaling mechanisms of NLRP3 upregulation by NMII C. burnetii, BMDM from mice deficient in TLR4, TLR2, TLR2 and 4, and the adaptor molecules, MyD88 and TRIF were infected and NLRP3 levels were measured by immunoblotting and compared to WT cells (Fig. 6). Cells from double knockout mice (Tlr2/4−/−) were employed to remove potential compensatory effects of TLR2 or TLR4. 50 NLRP3 levels in NMII-infected Tlr2−/− −, Tlr2/4−/−, and MyD88−/− BMDM were markedly reduced compared to WT BMDM indicating TLR2 and MyD88 are required for NLRP3 priming by NMII C. burnetii (Fig. 6). Furthermore, NLRP3 levels of infected cells from Tlr4−/− and Trif−/− mice were equivalent to those seen in WT cells, suggesting that these molecules are dispensable for NLRP3 upregulation by NMII C. burnetii (Fig. 6).
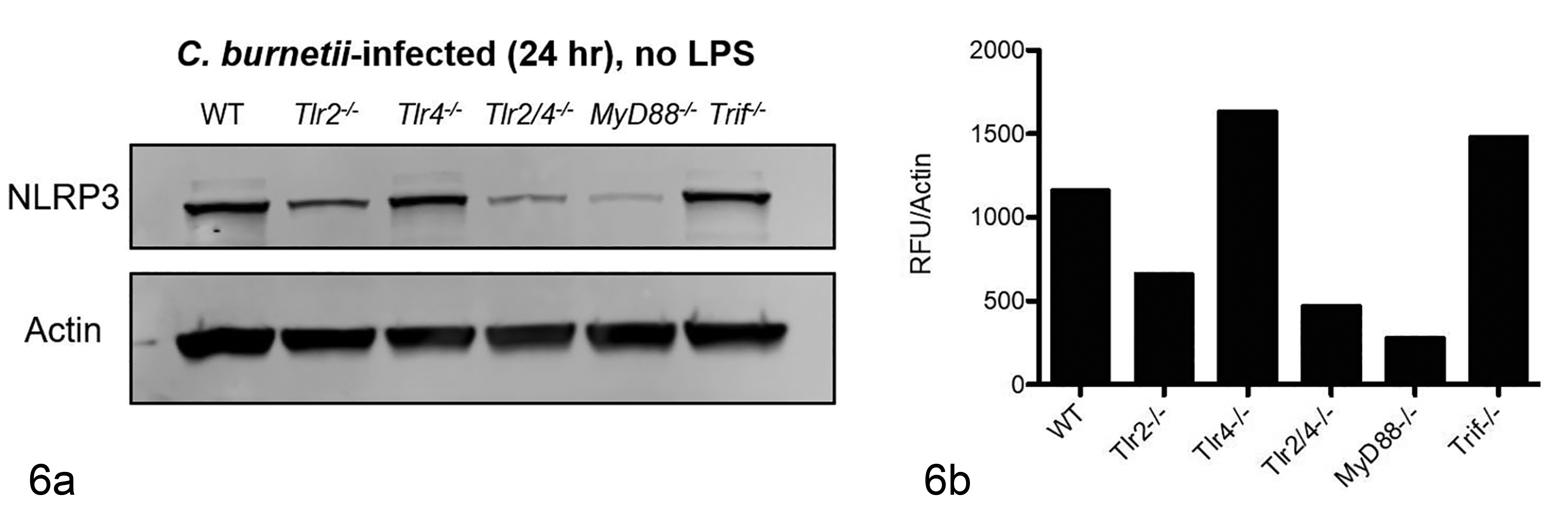
(a) In NMII C. burnetii–infected bone marrow–derived macrophages (BMDM), there is a marked reduction in NLRP3 levels in Tlr2−/− and MyD88−/− BMDM when compared to wildtype (WT) BMDM. TLR4 − and TRIF − BMDM lysates are similar to WT BMDM levels. (b) Representative densitometry and with normalization to actin highlight these differences, indicating MyD88 and TLR2 are involved in NLRP3 priming by NMII C. burnetii. Immunoblots and graphs are representative of at least 2 individual experiments.
NMII C. Burnetii Mutants Lacking the T4SS Do Not Cause Cell Death or IL-1β Secretion but Still Prime and Potentiate NLRP3 Inflammasomes
Since the T4SS of C. burnetii is required for inhibition of another form of programmed cell death (apoptosis), we evaluated the effect of T4SS deficiency on caspase-1 activation and NLRP3 inflammasome priming. BMDM from WT mice were infected with WT NMII C. burnetii or mutant NMII lacking the T4SS (▵dotA) or left uninfected. For NLRP3 priming experiments, BMDM were either LPS-treated or left untreated for the last 4 hours. Cell lysates were separated by SDS-PAGE and probed for NLRP3 and pro-IL-1β as described above. Compared to uninfected BMDM, those infected with WT NMII (Cb) and ▵dotA NMII had similarly robust upregulation of NLRP3 regardless of LPS stimulation (Fig. 7a, b). Similar to WT NMII, ▵dotA NMII induced modest increases in pro-IL-1β compared to NLRP3 (Fig. 7a, c). These findings suggest that priming of NLRP3 inflammasomes was not dependent on or affected by C. burnetii T4SS.
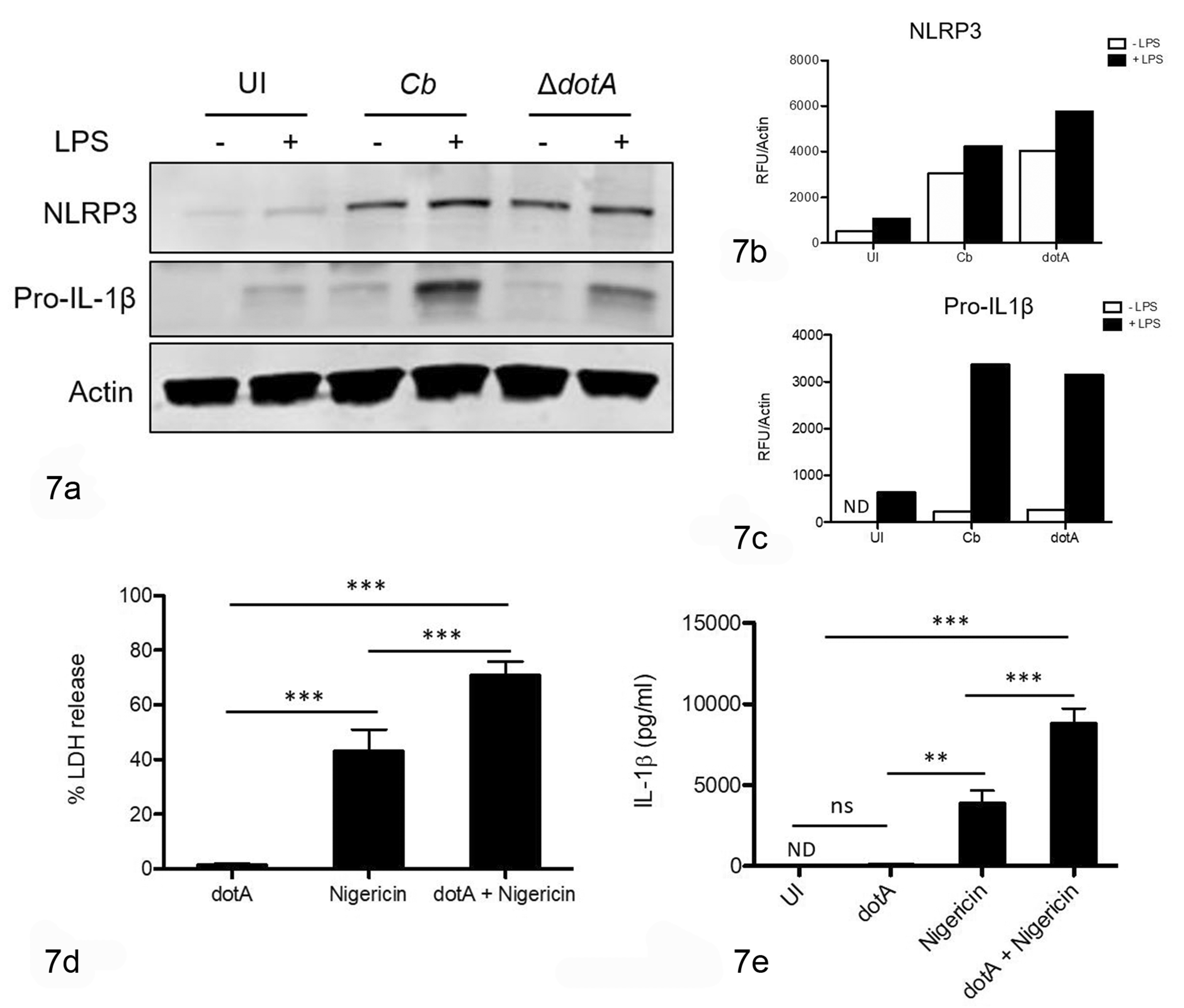
(a) Bone marrow–derived macrophages (BMDM) infected with ΔdotA NMII C. burnetii have similar NLRP3 and pro-IL-1β levels compared to wildtype NMII (Cb). With the addition of LPS, NLRP3 levels are increased in uninfected, NMII-infected, and ΔdotA-infected BMDM. In contrast, BMDM infected with NMII or ΔdotA have slightly increased levels of pro-IL-1β compared to uninfected BMDM, but less than uninfected BMDM treated with LPS. (b) Actin normalization of NLRP3 levels demonstrates the increases seen upon NMII infection (white bars) and the additive effect of LPS treatment (black bars). (c) Pro-IL-1β levels with normalization to actin are markedly increased following LPS treatment (black bars), with less robust induction by NMII and ΔdotA infections alone (white bars). (d, e) Wildtype BMDM were left uninfected or infected with ΔdotA and subsequently treated with nigericin, then cytotoxicity (d) and IL-1β secretion (e) were measured. BMDM infected with ΔdotA have minimal cytotoxicity (d) and IL-1β secretion (e). BMDM pre-infected with ΔdotA and then treated with nigericin have significantly increased cytotoxicity and IL-1β secretion compared to BMDM treated with nigericin alone. Data represent at least 2 (a-c) or 3 (d, e) individual experiments. Two-way ANOVA with Bonferroni’s multiple comparison test, (**) P < .001, (***) P < .0001, ns = not significant, ND = not detected.
Next, we assessed cytotoxicity and IL-1β secretion using nigericin treatment as a positive control and as a NLRP3 agonist to determine if (a) ▵dotA NMII induces pyroptosis and IL-1β secretion; (b) ▵dotA NMII potentiated these caspase-1-mediated effects similar to WT NMII. Cells infected with ▵dotA NMII did not exhibit cytotoxicity or secrete IL-1β (Fig. 7d, e). Furthermore, pre-infection with ▵dotA NMII resulted in significantly more pyroptosis and IL-1β secretion in nigericin-treated cells (Fig. 7d, e), thus indicating that the T4SS is not required for priming of or avoidance of cytosolic detection by NLRP3 inflammasomes.
Pulmonary NMII C. burnetii Infection Causes Transient Splenomegaly, Increased Bacterial Burdens and Macrophage Expansion in the Lungs, and Hepatic Microgranulomas in WT C57BL/6 Mice
An in vivo model was established using NMII C. burnetii to infect WT C57BL/6J mice via a pulmonary exposure over a 3-week time interval. Twelve- to 16-week-old male and female WT C57BL/6J mice were infected with NMII C. burnetii (1 × 107 bacteria) suspended in PBS via oropharyngeal instillation. Control mice were instilled with PBS only and were sacrificed at either 3- or 7-days post instillation. All mice were evaluated for clinical signs, body weight, spleen weight (as a percentage of body weight), pulmonary bacterial burdens, and pulmonary and hepatic macrophage infiltrates.
No mice showed clinical signs of disease or lost significant weight during the 3-week infections. At 3 days post infection (dpi), NMII-infected mouse lungs had increased alveolar macrophages with rare neutrophils and pyogranulomatous interstitial pneumonia with lymphocytes, plasma cells, and macrophages forming cuffs around bronchioles and vessels (data not shown). At 7 dpi, alveoli contained increased macrophages and flocculent eosinophilic (proteinaceous) fluid (Fig. 8b). In lungs from mice at 21 dpi, minimal cellular infiltrates were present (Fig. 8c). PBS-treated mice had no detectable histologic changes in the lungs (Fig. 8a). NMII C. burnetii were detectable via IHC by 3 dpi and most numerous at 7 dpi. when they found within macrophages scattered throughout the alveoli and interstitium (Fig. 9b). NMII C. burnetii were decreased and sparse by 21 dpi (Fig. 9c). Low numbers of NMII C. burnetii were detected in the spleens and livers of infected mice as early as 3 dpi via IHC, confirming bacterial dissemination from the lungs (data not shown). NMII C. burnetii were not detected in lungs, spleens, or livers of PBS-treated mice (Fig. 9a). F4/80-positive cells (macrophages) were most numerous in the lungs at 7 dpi (Figs. 10b, 12a) and decreased significantly by 21 dpi (Figs. 10c, 12a). By 3 dpi and most numerous at 7 dpi, livers of NMII-infected mice had multifocal microgranulomas, characterized as variably sized nodular aggregates of F4/80-positive macrophages with fewer lymphocytes, plasma cells, and neutrophils centered on rare necrotic hepatocytes (Figs. 11b, 12b). Hepatic microgranulomas increased in number and size up to 7 dpi (Fig. 11b) and then decreased by 21 dpi (Figs. 11c, 12b). Within the lung and liver of PBS-control mice, F4/80-labeled macrophages were limited to the perivascular and peribronchiolar regions and the reticuloendothelial framework, respectively (Figs. 10a and 11a).
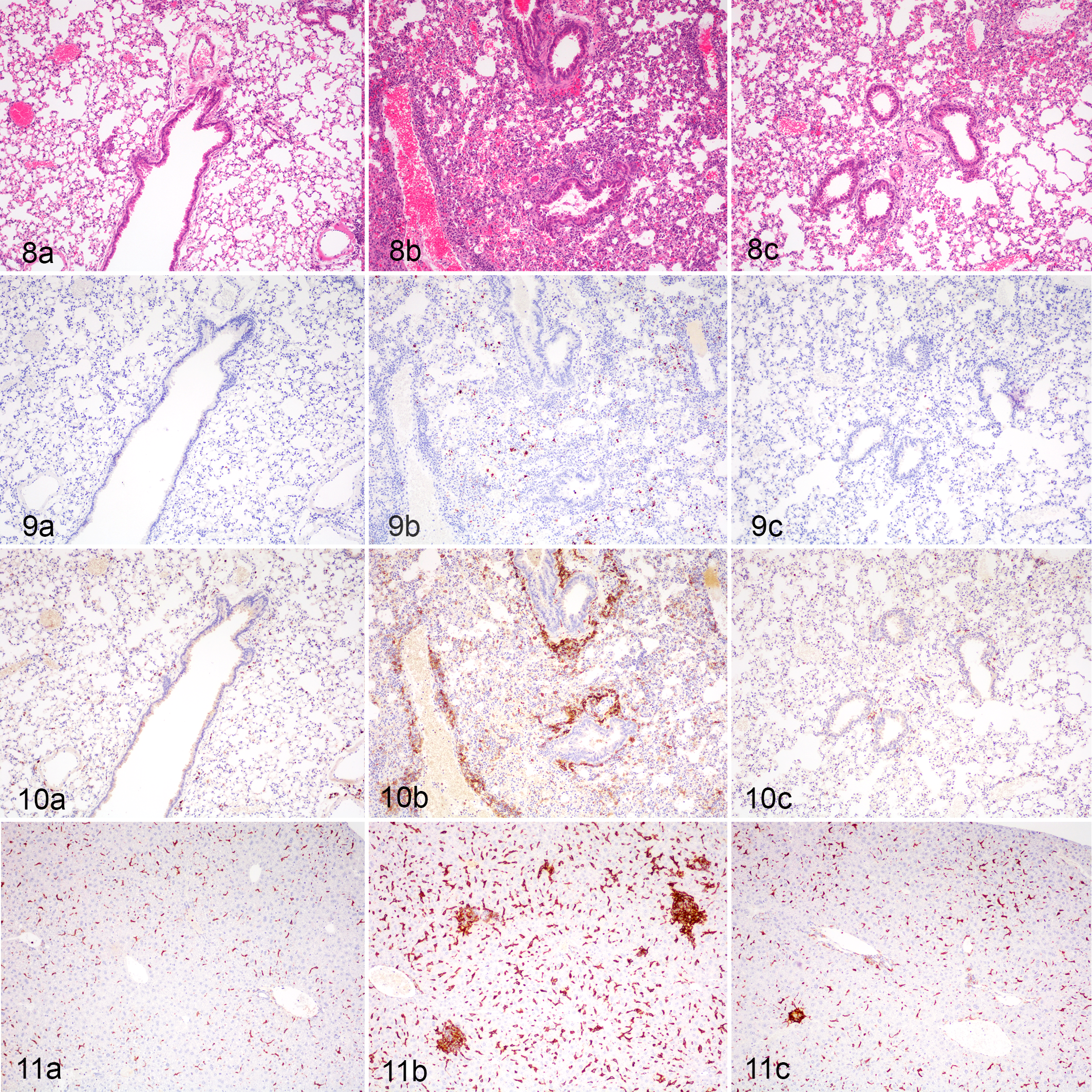
Coxiella burnetii Nine Mile II strain (NMII) or PBS-treated (control), lung, mice.
Detection and (semi)-quantification of NMII C. burnetii in the lungs was performed via qPCR (quantitative polymerase chain reaction; Fig. 12c) in addition to IHC (Figs. 9, 12d). Pulmonary burdens of NMII C. burnetii increased significantly by 3 dpi. By 7 dpi, detection of C. burnetii DNA decreased to baseline levels and remained very low on days 14 and 21 post infection (Fig. 12c). In contrast, immunolabeling of C. burnetii antigen continued to increase up to 7 dpi and afterwards declined significantly by 21 dpi (Fig. 12d).
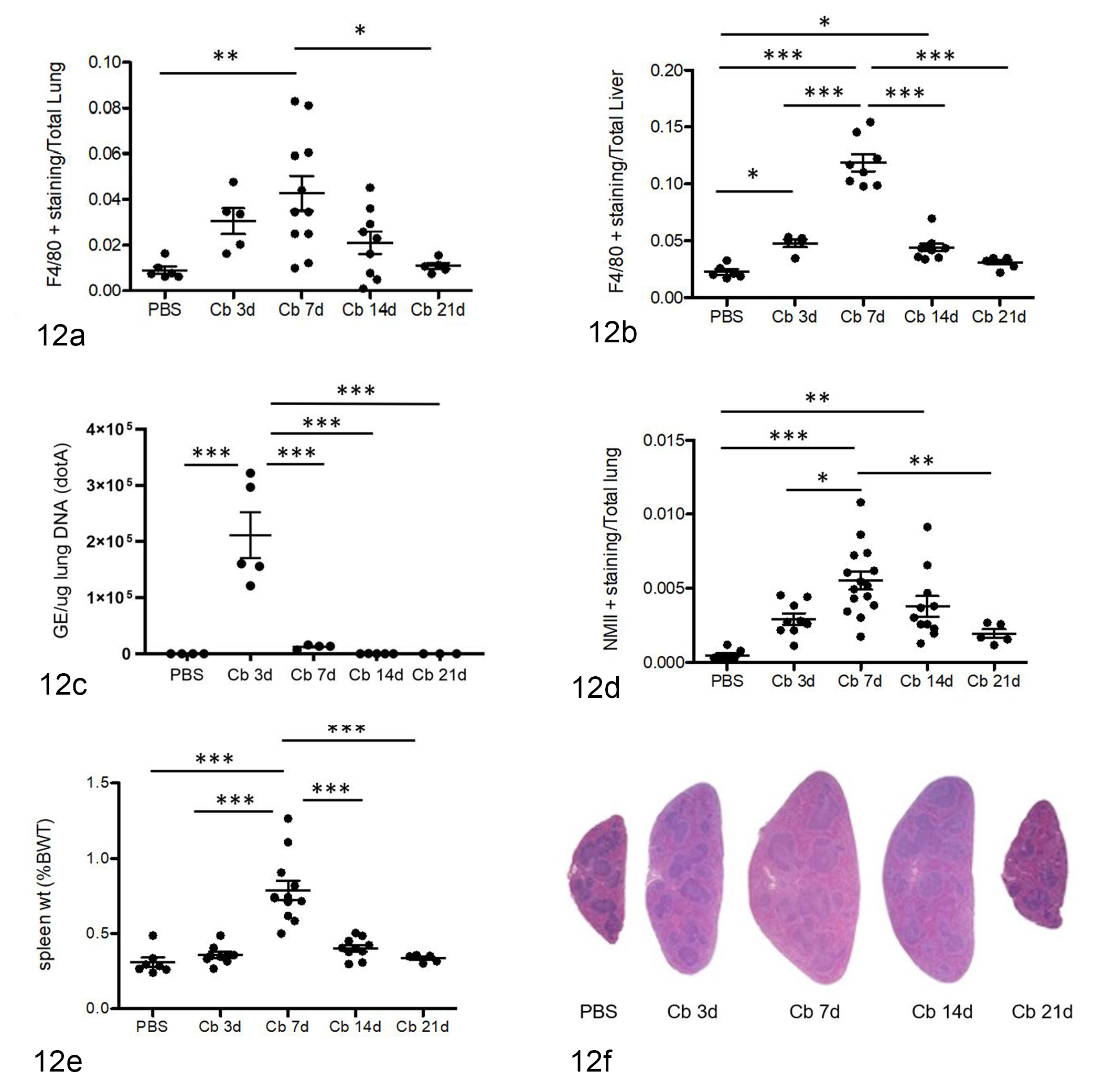
(a) Pulmonary macrophages (F4/80+) were significantly increased by 7 dpi with marked reduction by 21 dpi after challenge with Coxiella burnetii NMII strain (Cb). (b) Hepatic macrophages (F4/80+) had similar kinetics as for pulmonary macrophages with peak at 7 dpi and return to baseline (PBS) levels at 21 dpi. (c) Real-time PCR (qPCR) was performed on mouse lung extracts to quantify C. burnetii genomic DNA. Pulmonary bacterial burdens peaked at 3 dpi and were markedly reduced by 7 dpi. (d) Semiquantitative microscopy was performed on lung sections immunolabeled for C. burnetii NMII. NMII were detected in the alveolar macrophages by 3 dpi. In contrast to qPCR results immunolabeled NMII were significantly increased at 7 dpi, before decreasing at 21 dpi. (e, f) Splenomegaly was grossly evident at 3 dpi (f; Cb 3d), splenic weights were significantly elevated at 7 dpi, and subsequently declined to normal by 21 dpi. Each treatment group consisted of at least 4 or 5 mice. One-way ANOVA with Bonferroni’s multiple comparison test, (*) P < .05, (**) P < .001, (***) P < .0001.
In NMII-infected mice, spleen size increased by 3 dpi, peaked at 7 dpi, and subsequently returned to the size of PBS-treated mice by 21 dpi (Fig. 12e, f). Histologically, splenomegaly was predominantly attributed to expansion of the red pulp with increased macrophages and fewer hematopoietic cells. In spleens of NMII-infected mice at 7 dpi and 14 dpi, lymphoid follicles and periarteriolar sheaths were also enlarged with prominent mantle zones (Fig. 12f).
Together, these data demonstrated that C57BL/6 J WT mice, although considered clinically resistant to NMII C. burnetii, in fact develop consistent multi-organ histologic lesions and splenomegaly following pulmonary infection. Via molecular quantification, pulmonary bacterial burdens were highest at 3 dpi although infected macrophages were most numerous in histologic sections at 7 dpi. Furthermore, NMII C. burnetii were able to disseminate systemically from the lungs to spleen and liver.
NLRP3 and Caspase-1 Are Dispensable to the Resolution of Splenomegaly, Hepatic Microgranulomas, and Pulmonary Bacterial Burdens Following Pulmonary Infection with NMII C. burnetii
To determine the biological relevance of NLRP3 inflammasome activation to NMII C. burnetii pathogenesis, we compared WT mice and mice deficient in NLRP3 or caspase-1 using our in vivo model described above (Fig. 13). We evaluated WT, Nlrp3−/ −, and Casp1−/ − infected and PBS-control mice over a 3-week period following oropharyngeal instillation of NMII C. burnetii.
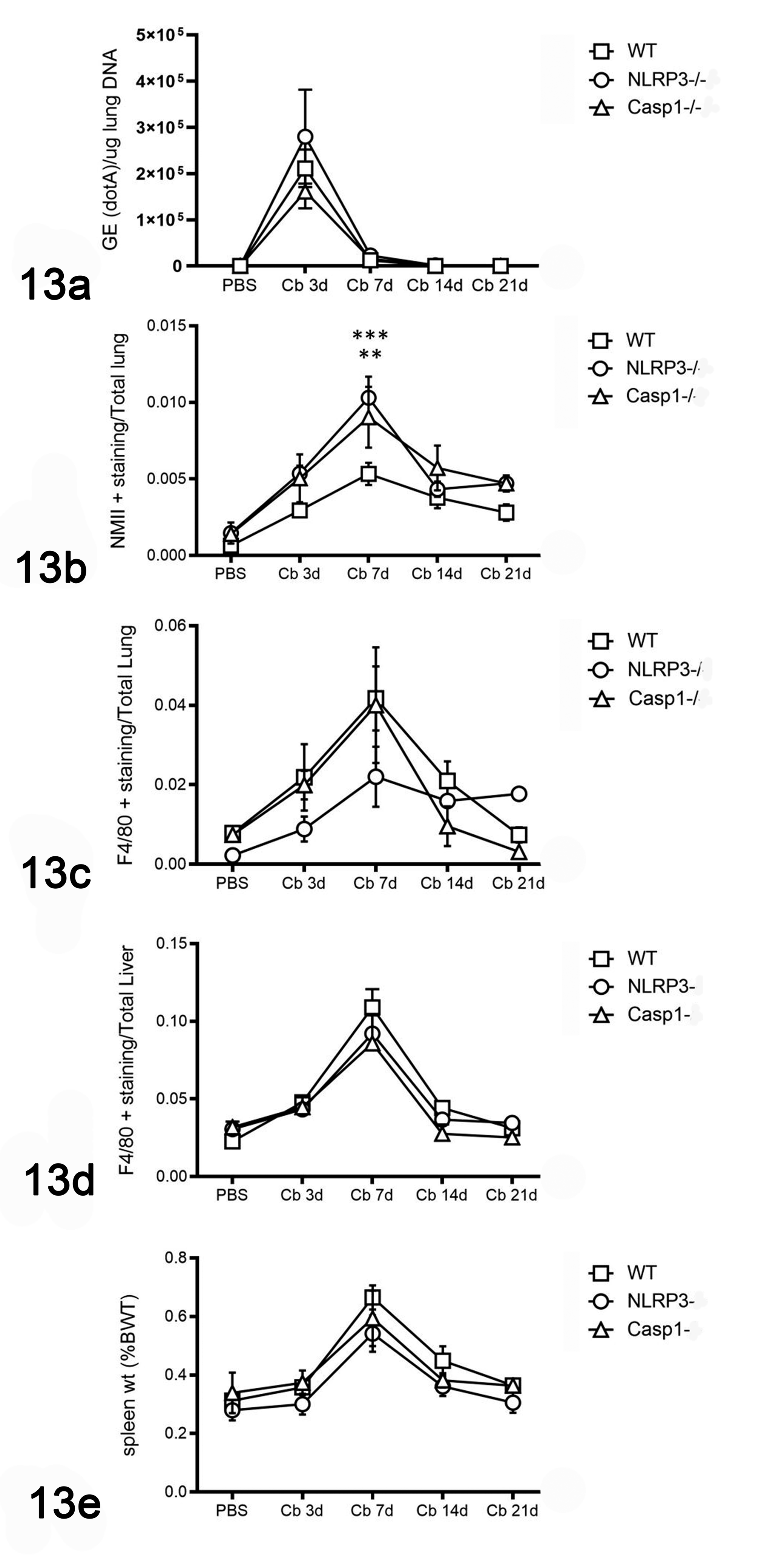
(a) Pulmonary Coxiella burnetii NMII (Cb) bacterial burdens measured by qPCR peaked at 3 days post infection (dpi) and were not significantly different between wildtype (WT), Nlrp3−/− and Casp1−/− mice. (b) Based on IHC for Coxiella burnetii, pulmonary bacterial burdens were significantly higher in Nlrp3−/− and Casp1−/− than WT mice; however, they were not significantly different at any other time points. (c) Macrophages within the liver increased significantly by 7 dpi in all mice. There was no difference in hepatic macrophages between WT mice, Nlrp3−/− and Casp1−/− mice over the 21-day infection interval. (d) Pulmonary macrophages showed similar trends as for pulmonary bacterial burdens, although there was no significant difference between WT mice, Nlrp3−/− and Casp1−/− mice. (e) Development and resolution of splenomegaly as measured by percentage of body weight was similar in all mice over the 21-day infection interval. Each treatment group consists of at least 4 mice. Two-way ANOVA with Bonferroni’s multiple comparison test, (**) P < .05, (***) P < .001.
None of the Nlrp3−/− and Casp1−/− − mice had clinical signs or significant weight loss. Lung and liver microscopic lesions of Nlrp3−/− and Casp1−/− mice were also like those in WT mice. By molecular detection of C. burnetii genomic DNA in lung extracts, there was no significant difference between WT and knockout mice (Fig. 12a). Interestingly, at 7 dpi, NMII-positive immunolabeling in the lungs was significantly higher in Casp1−/− (P < .05) and Nlrp3−/− (P < .001) mice; however, by 21 dpi, there was no significant difference detected (Fig. 13b). F4/80-positive immunolabeling in the lungs was not significantly different between WT and knockout mice at any time points (Fig. 13c). Macrophage abundance in the liver followed a virtually overlapping trend in the knockout mice compared to WT mice (Fig. 13d). Splenomegaly of Nlrp3−/−and Casp1−/− mice followed the same temporal trend as for WT mice (Fig. 13e). Furthermore, subgrossly and histologically, spleens from Nlrp3−/− and Casp1−/− mice were indistinguishable from WT mice at 3, 7, 14, and 21 dpi (Figs. 12f, 13e). Together, these data suggest that NLRP3 and caspase-1 are dispensable to macrophage responses in the lung and liver, to the eventual decreases in pulmonary bacterial burdens, and to the development and resolution of splenomegaly following NMII C. burnetii pulmonary infection in C57BL/6J mice.
Discussion
Recent epizootics in ruminant reservoirs and human populations have rejuvenated research efforts to better understand the molecular mechanisms underlying the complicated and dynamic host-pathogen interactions that occur during Q fever. 12,41,78 Here, we examined the interactions between a highly adapted obligate intracellular bacterium, Coxiella burnetii (Nine Mile Phase II), and a conserved innate defense mechanism, the inflammasome.
We first established an in vitro model using the clinically resistant C57BL/6 mouse as a source for bone marrow–derived macrophages (BMDM), a cell type considered restrictive to NMII C. burnetii. Our findings confirmed that NMII C. burnetii infect and survive up to 72 hours in C57BL/6J BMDM in vitro with evident viable intracellular bacteria via fluorescence microscopy. We observed no evidence of cell death, caspase-1 activation, or substantial IL-1β secretion despite the presence of viable intracellular bacteria. Thus, we confirmed that NMII C. burnetii does not induce caspase-1 activation or IL-1β secretion in mouse BMDM as was reported in recent studies. 13,28
C. burnetii lacks flagellin but has a T4SS that secretes bacterial proteins and products into the cytosol, so we focused on interactions between C. burnetii and the NLRP3 inflammasome. 23,34 We hypothesized that C. burnetii avoids detection by the receptors involved in NLRP3 inflammasome formation and activation, which include those at the cell membrane (TLRs) and cytosolic sensors, specifically NLRP3 and the adaptor protein ASC. Lack of NLRP3: ASC foci formation suggests that NMII C. burnetii is not detected by NLRP3 in host cell cytosol. Without NLRP3 sensing and subsequent ASC foci formation, there is no recruitment and activation of caspase-1 or the downstream effects of pyroptosis and IL-1β secretion. Thus, the activation of NLRP3 by sensing of a PAMP or DAMP (signal 2) during C. burnetii infection of BMDM is either avoided or actively inhibited.
To determine if NMII C. burnetii can actively inhibit inflammasomes, we used potent agonists of the NLRP3 and NLRC4 inflammasomes in BMDM pre-infected with NMII (Fig. 4). Surprisingly, we found that NMII was unable to attenuate inflammasome formation and instead NMII-infected BMDM had higher cytotoxicity and IL-1β secretion. This inability to attenuate inflammasome activation corroborates results from previous studies utilizing other agonists including ATP following LPS stimulation, nigericin for activation of NLRP3 inflammasomes, and Legionella pneumophila for induction of NLRC4 inflammasomes. 13,28 It has also been demonstrated that C. burnetii NMII is unable to inhibit the AIM2 inflammasome induced by dsDNA. 28 Collectively, these data indicate that C. burnetii does not attenuate or inhibit inflammasome activation at the level of the effector protease, caspase-1, or downstream of caspase-1 activation. Our novel finding of inflammasome potentiation by NMII C. burnetii warranted investigation into possible mechanisms. Based on previous reports, we hypothesized NMII C. burnetii provided PAMP signaling to BMDM resulting in NLRP3 inflammasome priming and subsequent increases in cytotoxicity and IL-1β secretion. 7,89 Notably, NMII C. burnetii has a large chromosomal deletion in LPS rendering it avirulent compared to NMI and other described strains, thus other PAMPs and Toll-like receptor signaling pathways were considered. 22,89
Through immunoblotting, we found that NMII C. burnetii robustly upregulates NLRP3 and to a lesser extent pro-IL-1β in the absence of LPS (Fig. 5). Interestingly, NMII-infected BMDM had higher levels of NLRP3 following LPS treatment, which indicates an additive effect. Furthermore, LPS was shown to be an effective agent for the induction of pro-IL-1β. This priming resulted in the potentiation of caspase-1-mediated effects when NMII-infected BMDM were subsequently treated with the potent NLRP3 agonists, nigericin and Yptb. Also surprising was the potentiation noted in Stm-infected BMDM pre-infected with NMII. It is well know that some pathogenic bacteria can stimulate more than one inflammasome, highlighting the importance of inflammasomes to the innate response during microbial infections. 18,21,55,58,67,81 Specifically, NLRC4 and NLRP3 inflammasomes may have redundant roles in response to Salmonella enterica serovar Typhimurium depending on bacterial flagellin up- or downregulation and length of infection. 18,55,67 To elucidate the mechanism of NMII C. burnetii inflammasome potentiation during co-infections with Stm, we measured NLRC4 levels in NMII-infected BMDM and found that NLRC4 levels were mildly increased as for uninfected LPS-treated BMDM. This suggests that although not required for activation or priming, treatment with LPS (and other PAMPs such as lipopeptides) does promote NLRC4 inflammasomes and this is likely the reason for the potentiation noted in Stm-infected BMDM pre-infected with NMII.
By utilizing BMDM from genetically modified mice deficient in TLRs (TLR2, TLR4, TLR2/4) and adaptor molecules (MyD88, TRIF), we determined that NLRP3 priming by NMII C. burnetii is dependent on TLR2-MYD88 signaling and independent of the TLR4-TRIF pathway (Fig. 6). This was an interesting finding in that there are conflicting theories as to the importance of TLR2 and TLR4 for the detection of C. burnetii. 2,65,68,69,89 Host detection and responses to Nine Mile C. burnetii through TLR-signaling are further complicated as a result of LPS or phase variation, with variable engagement and roles of TLR2 and TLR4 depending on LPS structure. 7,37,89 Our results indicate NMII with its truncated LPS does not engage host TLR4 or TRIF and instead interacts with TLR2 and the adapter protein MyD88. Although TLR2-mediated detection of NMII C. burnetii results in the upregulation of NLRP3, it does not lead to inflammasome formation and subsequent caspase-1 activation. The lack of caspase-1 activation has been described previously in different cell types including mouse alveolar macrophages and bone marrow–derived macrophages, the latter of which were used in this study. 13,28 Importantly, mutant C. burnetii lacking the T4SS (▵dotA) did not induce caspase-1 activation, IL-1β secretion, or cell death despite robustly priming NLRP3 inflammasomes. This lack of inflammasome activation was seen in macrophages infected with mutant bacteria lacking the recently discovered IcaA (inhibition of caspase activation) protein produced by C. burnetii. 28 IcaA attenuates the activation of caspase-1 that is triggered through the non-canonical caspase-11-mediated pathway by an unknown mechanism but does not act upon caspase-1 directly. 28 Recent studies have linked non-canonical inflammasome activation, mediated by caspase-11 in mice and caspase-4 and -5 in humans, to canonical NLRP3 inflammasomes regulated by caspase-1, during gram-negative sepsis and when bacterial pathogens access the host cytosol. 17,22,70 Although the exact mechanisms are unknown, type 1 interferon signaling and the adaptor protein TRIF are involved resulting in caspase-4/11 activation, subsequent pore formation, changes in ionic balances, detection by NLRP3 sensors, inflammasome formation, and caspase-1 activation. 22,70 Our data show that TRIF is not required for NLRP3 priming by NMII C. burnetii. Furthermore, our findings suggest that the lack of caspase-1 activation by C. burnetii is not reliant on T4SS or a specific effector, and rather lack of cytosolic detection by NLRP3 of the bacteria and its products. Although the probability of one of the 130 known C. burnetii effector proteins being sensed seems considerable, collectively our data and those from related studies suggest C. burnetii effectively avoid cytosolic detection in a T4SS-independent manner negating the NLRP3 inflammasome as a player in the initial immune response. 13,28
Interestingly, B1a B cells derived from the peritoneal cavity of mice exhibited caspase-1-mediated cell death (pyroptosis) following infection with NMII C. burnetii. 74 This subset of B cells was previously found to be important to the immune response to C. burnetii. The authors deduced that NMII-induced caspase-1 dependent pyroptosis may require the T4SS, TLR2 signaling, and NLRP3 inflammasomes. 74 Other cell types have been shown to behave differently following C. burnetii infection with divergent cell death responses depending on bacterial phase variation and the cell’s activation states, in cell types including human monocytes, monkey and mouse alveolar macrophages, and THP-1 cells (human monocyte cell line). 19,84,85,91 Clearly, host-pathogen interactions are specific to certain cell types and there are overlapping mechanisms across immune cells to ensure a successful collective response to the most highly adapted of pathogens.
To better examine interactions between NMII C. burnetii and host inflammasomes and to elucidate the biological relevance of these immune platforms, we utilized an in vivo model, which encompasses entire organ systems, various immune cell types, and a longer course of infection following a natural route of infection. With our in vivo model, we found that despite no outward signs of illness or weight loss, C57BL/6J mice develop pulmonary, hepatic, and splenic lesions with bacterial dissemination following pulmonary exposure of NMII C. burnetii. Although C57BL/6 mice are considered clinically resistant to NMII C. burnetii, it is clear that immunocompetent (wildtype) mice can develop similar lesions to human Q fever patients and the gold-standard animal model, guinea pigs, following infection with NMI and other virulent strains of C. burnetii. 4,77,88 In WT mice, following a similar trajectory as determined by histology and confirmed with immunohistochemical labeling of F4/80+ macrophages, pneumonia, and hepatic microgranulomas increased in severity up to 7 days post infection (dpi) and resolved by 21 days. We also found that the spleens of infected WT mice increased in size up to 7 dpi and resolved to normal size by 21 dpi. Splenomegaly was also reported in previous in vivo studies following intraperitoneal infection of immunocompromised mice. 65,79 Contributing to splenomegaly in our study were extensive macrophage infiltrates and increased extramedullary hematopoiesis, the latter of which is a common finding in mice following a systemic inflammatory response or antigenic stimulation.
Interestingly, using molecular quantification of NMII C. burnetii, we measured highest pulmonary burdens at 3 dpi compared to 7 dpi as determined using IHC. The discrepancy in time of the highest measured bacterial burdens in the lung was considered the result of the sensitivity of the assay and/or the half-life of the target (DNA versus protein).
Pulmonary infections were induced in mice deficient in NLPR3 or caspase-1 to determine the role of these inflammasome components in the resistance of C57BL/6J mice to NMII C. burnetii. The kinetics of macrophages in the lung, liver, and spleen, and pulmonary bacterial burdens, determined by both qPCR and IHC, were similar across WT, Nlrp3−/− and Casp1−/− mice following infection with NMII C. burnetii.
Notably, we found a statistically significant increase in bacterial burdens in mice deficient in NLRP3 and caspase-1 compared to WT mice at 7 dpi as measured by immunohistochemical labeling but not by qPCR for bacterial quantification. Besides assay differences, another possible mechanism for this dichotomy is the presence of NLRP3 agonists in the lungs during peak inflammation and microbicidal activity in response to C. burnetii. It is likely that there is some level of cell death within the lungs and release of potential NLRP3 agonists (ie, DAMPs) that provide Signal 2 for inflammasome and caspase-1 activation. Yet another possibility is the role of non-canonical inflammasomes, mediated by caspase-11 in mice, in the response to C. burnetii as well as its interplay with NLRP3 inflammasomes and caspase-1. 22 C. burnetii has a T4SS effector protein, IcaA, that can attenuate caspase-11 activation, thus mitigating NLRP3-mediated caspase-1 activation. 28 However, to date, there are no in vivo studies utilizing IcaA-deficient C. burnetii to elucidate its role during natural infections. Notably, the caspase-1-deficient mice used in our studies were also deficient in caspase-11, so the differences we see in pulmonary burdens at 7 dpi between WT and KO mice could be a result of the lack of either caspase-11 and or caspase-1. For our purposes, this in vivo model demonstrated that deficiency of NLRP3 or caspase-1 does not affect the ability of C57BL/6J mice to develop an inflammatory response and reduce pulmonary bacterial burdens following pulmonary infection with C. burnetii. Furthermore, NLRP3 and caspase-1 are not critical to the systemic responses to C. burnetii as evidenced by the nearly identical profiles of splenomegaly and hepatic macrophages.
In conclusion, these studies provided insight into innate immune responses to NMII C. burnetii and this bacteria’s interactions with NLRP3 inflammasomes. Our findings further suggest NLRP3 priming and subsequent activation are tightly controlled and bacteria possibly contribute to this regulation. Macrophage activation states are critical to immune responses during Q fever and susceptibility to C. burnetii infection. 8,25,57 Macrophage activation is also important to the individual cell’s survival as this relates to caspase-mediated cell death. 9 Considering C. burnetii can persist in tissues for months and even years during chronic Q fever and that there is dysregulation of inflammatory cytokines in these patients, determining the effect of macrophage activation states on NLRP3 upregulation could be enlightening. 38,41,75 By using C57BL/6J mice, which are M1 (Th1)-biased and clinically resistant to NMII C. burnetii, these findings may be more easily translated to those acute Q fever patients who successfully clear bacteria during the initial infection period.
Future studies utilizing other inbred mouse strains, such as BALB/c, which have M2 (Th2)-biased macrophages, or collaborative cross mice, will be beneficial to the understanding of complex host-pathogen interactions occurring in patients with acute and chronic forms of the disease. 11,41,64 In addition, completing these in vitro and in vivo experiments with NMI and other virulent strains under BSL3 conditions is warranted. A better understanding of the mechanisms required for early clearance of C. burnetii may lead to the development of immune therapies for chronic Q fever patients. Specifically, our in vivo model will be useful to study other important players in Q fever pathogenesis, such as TLR-signaling pathways and adaptor molecules, cytokines and cytokine receptors, T helper cells and the adaptive immune response, virulence of C. burnetii strains, and potentially novel therapeutic, diagnostic, and preventative strategies.
Supplemental Material
Supplemental Material, Combined_supplemental_materials-Delaney_et_al - Avoidance of the NLRP3 Inflammasome by the Stealth Pathogen, Coxiella burnetii
Supplemental Material, Combined_supplemental_materials-Delaney_et_al for Avoidance of the NLRP3 Inflammasome by the Stealth Pathogen, Coxiella burnetii by Martha A. Delaney, Andreas den Hartigh, Samuel J. Carpentier, Timothy P. Birkland, Donald P. Knowles, Brad T. Cookson and Charles W. Frevert in Veterinary Pathology
Footnotes
Acknowledgements
The authors would like to thank the members of the Cookson Laboratory in particular Matthew Johnson for assistance with mice and Dr Wendy Loomis for her insights and suggestions. We thank Dowon An for her technical expertise and assistance with in vivo studies. We thank Megan Larmore, Julie Hahn, and Brian Johnson of the Histology and Imaging Core for sample preparation and quantitative analysis. Special thanks to thesis committee members Drs Piper Treuting and Deborah Fuller for their guidance. Thanks to Dr Robert Heinzen, Diane Cockrell, and Dr Paul Beare at Rocky Mountain Laboratories who provided the expertise, training, and C. burnetii strains used for our studies. Thank you Devin Ruddick for assistance with figure editing.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Microscopy was performed at the W. M. Keck Microscopy Center with support of G. Martin and Nathaniel Peters and with support of NIH Award S10OD016240. This work was funded by USDA Grants 59-2090-5-004 and R01AI136468.
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.