
Abstract
An 11-year-old castrated male Chihuahua dog was presented with complaints of polyuria, polydipsia, abdominal enlargement, and alopecia. Hyperadrenocorticism was diagnosed on the basis of clinical signs, blood tests, adrenocorticotropin-stimulation test results, and an elevated serum adrenocorticotropin concentration. Contrast-enhanced magnetic resonance imaging showed that the pituitary gland was enlarged, compatible with a pituitary macroadenoma. Pituitary-dependent hyperadrenocorticism was suspected, and transsphenoidal hypophysectomy was thus performed for complete resection of the tumor. After surgery, the serum adrenocorticotropin concentration normalized and the hyperadrenocorticism resolved. Histological and immunocytochemical analyses revealed a benign tumor composed of mature neuronal cells and glial cells, suggestive of a ganglioglioma with immunolabeling for adrenocorticotropin. Careful analysis of the resected tumor revealed no pituitary adenoma tissue. The clinical and histopathologic findings indicated that the ganglioglioma was directly responsible for the hyperadrenocorticism. This is the first case of hyperadrenocorticism caused by a ganglioglioma in a dog.
Spontaneous hyperadrenocorticism is an endocrine disorder that results from excessive cortisol secretion by the adrenal cortex. Approximately 80% to 85% of dogs with hyperadrenocorticism have pituitary-dependent hyperadrenocorticism (PDH) caused by an adrenocorticotropin (ACTH)–secreting corticotroph adenoma, and 15% to 20% have adrenocortical-dependent hyperadrenocorticism caused by a glucocorticoid-secreting adrenocortical tumor. 9 Hyperadrenocorticism is rarely caused by ectopic secretion of ACTH 3,6,11 or aberrant receptor expression in the adrenal cortex. 10 Furthermore, pituitary ganglion cell tumors are reportedly a rare cause of PDH in humans. 8 Pituitary ganglion cell tumors are benign tumors showing neuronal differentiation and are categorized as either gangliocytomas or gangliogliomas. These tumors are either associated with a hormone-secreting pituitary adenoma or, rarely, the tumors secrete the hormones themselves. 7,8
In veterinary clinical medicine, the increasing prevalence of advanced imaging modalities has enabled detailed visualization of the pituitary and, in combination with endocrinological tests, has enabled accurate diagnosis of PDH. PDH is a common endocrine disease in dogs, but to our knowledge, pituitary ganglion cell tumors have not been reported. We herein report a canine case of an ACTH-secreting pituitary ganglioglioma that presented with a clinical picture of PDH.
An 11-year-old castrated male Chihuahua dog weighing 2.8 kg was referred to the Veterinary Medical Teaching Hospital of Nippon Veterinary and Life Science University with complaints of polyuria, polydipsia, abdominal enlargement, and alopecia. Hyperadrenocorticism had been diagnosed on the basis of clinical signs, blood test results, and ACTH stimulation test results at a private hospital. The basal (0-minute) serum cortisol concentration was 9.4 μg/dl (reference range, 0.5–6.0 μg/dl), and the serum cortisol concentration 60 minutes after intravenous administration of 0.25 mg synthetic ACTH (Cortrosyn; Daiichi Sankyo, Tokyo, Japan) was 52.6 μg/dl (reference range, 6.0–22.0 μg/dl). The dog had been treated with trilostane for 1 year before the referral.
At the first visit to our institution, abdominal radiography revealed hepatomegaly. Routine laboratory examination showed an increased serum alkaline phosphatase concentration (3241 U/L; reference range, 47–254 U/L). Other serum biochemical variables were within the reference ranges. The plasma ACTH concentration and serum total thyroxine, free thyroxine, and thyroid-stimulating hormone concentrations were assayed as previously described. 18 The basal plasma ACTH concentration was 232 pg/ml (reference range, 6.0–58.0 pg/ml). The serum total thyroxine, free thyroxine, and thyroid-stimulating hormone concentrations were 6.4 nmol/L (reference range, 13.0–51.0 nmol/L), 13.9 pmol/L (reference range, 6.6–40.0 pmol/L), and 0.25 ng/ml (reference range, 0.03–0.38 ng/ml), respectively. Abdominal ultrasonography revealed bilateral enlargement of the adrenal glands (left adrenal, 8.1-mm diameter; right adrenal, 8.3-mm diameter).
Imaging of the pituitary gland was performed under anesthesia using a 3.0-T superconducting magnet (Signa HD; GE Healthcare Japan, Tokyo, Japan). T1-weighted transverse and sagittal images were obtained using the gradient-echo method with a 7-ms repetition time, 3-ms echo time, and 2.0-mm-thick consecutive slices before and after an intravenous bolus injection of the contrast medium gadodiamide hydrate (Omniscan; Daiichi Sankyo, Tokyo, Japan) at 0.05 mmol/kg body weight. Contrast-enhanced magnetic resonance imaging showed that the pituitary gland was enlarged (height, 10.1 mm; pituitary height/brain area ratio, 0.70 × 10–2 mm–1) (Fig. 1). 14 A pituitary macroadenoma was suspected, and transsphenoidal hypophysectomy was performed as described previously on the 28th day after the first visit. 16
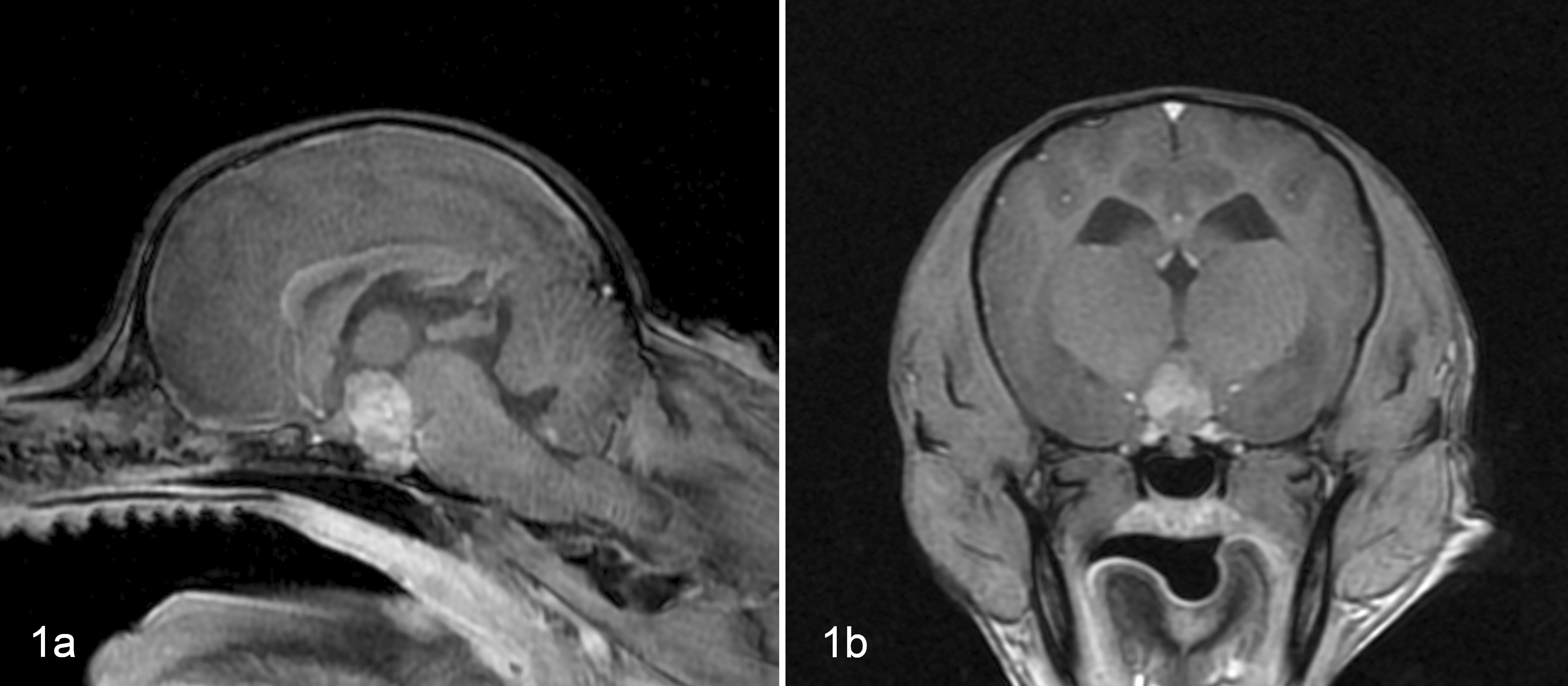
Pituitary tumor, dog. Contrast-enhanced magnetic resonance T1-weighted images of the pituitary gland at the time of diagnosis of pituitary-dependent hyperadrenocorticism. A mass is present in the pituitary region and filled the third ventricle. (a) Sagittal image. (b) Transverse image.
The resected tumor tissue was fixed in 4% paraformaldehyde, embedded in paraffin, and processed for histological examination. Sections (2 μm thick) were stained with hematoxylin and eosin. Immunohistochemical staining was performed by the peroxidase-labeled antibody method using monoclonal mouse anti–synthetic human ACTH (1:200; Dako Japan, Kyoto, Japan), 18 monoclonal mouse anti–synthetic human thyroid-stimulating hormone (1:100; Advanced ImmunoChemical, Long Beach, CA), 18 polyclonal rabbit anti–human growth hormone (1:400; Dako Japan), 2 polyclonal rabbit anti–human prolactin (1:400; Dako Japan), 2 polyclonal rabbit anti–human arginine vasopressin (1:400; Sigma-Aldrich, St Louis, MO), 17 polyclonal rabbit antisynthetic human corticotropin-releasing hormone (CRH) (1:400; Abcam, Tokyo, Japan), rabbit anti–bovine glial fibrillary acidic protein (1:400; Dako Japan), 1 monoclonal mouse anti–human neuron-specific enolase (1:400; Dako Japan), 1 and monoclonal mouse anti–human neurofilament protein (1:400; Dako Japan). 19 Horseradish peroxidase (HRP)–conjugated F(ab′)2 fragments of sheep anti–mouse IgG or donkey anti–rabbit IgG (Amersham Pharmacia Biotech, Piscataway, NJ) were used as secondary antibodies for pituitary hormones. Polymer-HRP-conjugated anti–rabbit IgG or anti–mouse IgG antibody (Dako Japan) was used as a secondary antibody for hypothalamic hormones and neuronal markers. Deparaffinized sections were treated with 0.3% hydrogen peroxide in methanol for 30 minutes to block endogenous peroxidase activity and incubated with the primary antibody overnight at 4°C. Next, the slides were incubated for 1 hour at room temperature with secondary antibody. Immunoreactivity was visualized using 50 mM Tris-HCl buffer (pH 7.6) containing 0.01% 3,3-diaminobenzidine tetrahydrochloride, 0.005% hydrogen peroxide, and 0.01% sodium nitrate, followed by a counterstain with hematoxylin. Normal dog pituitary or hypothalamus specimens were used as positive controls and showed appropriate immunolabeling. Negative controls were obtained by substituting the primary antibodies with normal buffer solution. To verify the specificity of the immunoreaction for CRH, reabsorption of the CRH antibody with synthetic human CRH (10−6 M; Peptide Institute, Osaka, Japan) was used for immunolabeling of normal dog hypothalamus tissue. This control was negative, whereas positive immunoreaction was observed using the same CRH antibody without reabsorption.
Pathological examination of the resected tumor demonstrated benign neoplastic tissue. The tumor had dysplastic neuronal cells and spindle cells with a glial-like fibrillary matrix (Fig. 2). No mitotic activity was seen, and the cells demonstrated only mild pleomorphism. In some areas, large dysplastic neuronal cells showed a ballooned appearance and binucleation (Fig. 3). These findings along with the presence of strong cytoplasmic immunoreaction for neurofilament protein and neuron-specific enolase in the neuronal cells (Fig. 4, 5) and glial fibrillary acidic protein in the glial cells (Fig. 6) led to the diagnosis of ganglioglioma. Immunohistochemistry for pituitary and hypothalamic hormones revealed moderate to strong cytoplasmic ACTH immunoreactivity in neuronal cells (Fig. 7) and negativity for thyroid-stimulating hormone, growth hormone, prolactin, arginine vasopressin, and CRH. Careful analysis of all resected tissue performed on serial sections at 30-μm intervals did not reveal any findings consistent with a pituitary adenoma.
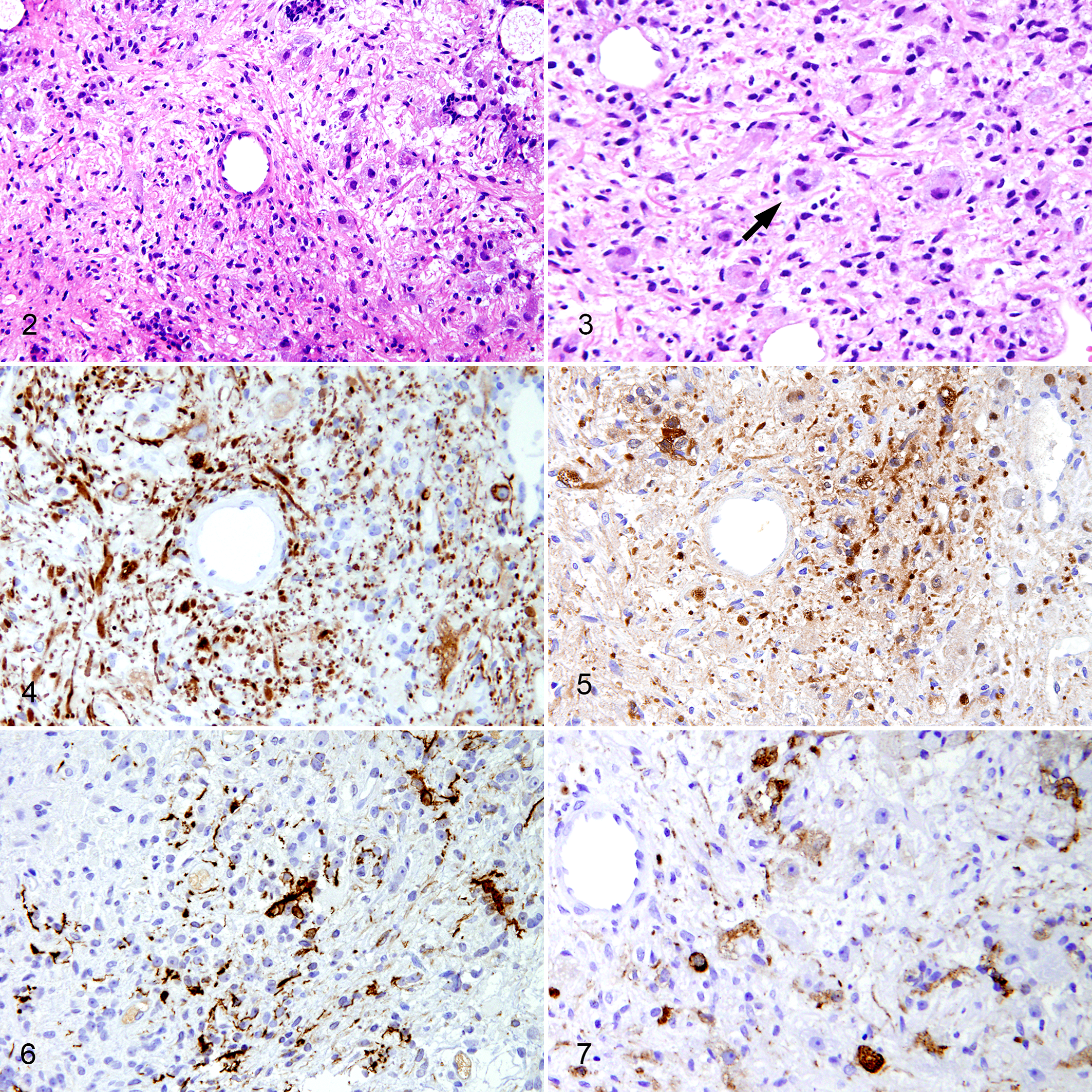
Ganglioglioma, pituitary gland, dog.
After a successful surgery and uneventful recovery, the dog was released from the hospital with hormone replacement therapy consisting of cortisone acetate and thyroxine as previously described. 16 One week after surgery, the basal ACTH concentration was 16.4 pg/ml. Four weeks after hypophysectomy, the basal ACTH concentration was 13.5 pg/ml, and the basal and post-ACTH serum cortisol concentrations were 0.6 and 6.4 μg/dl, respectively. These results indicated successful hypophysectomy and remission of the hypercortisolemia. At the time of the last follow-up 22 months after surgery, the dog was still alive and free of signs of hyperadrenocorticism.
According to the International Histological Classification of Tumors in Domestic Animals by the World Health Organization, intracranial neuronal and mixed neuronal–glial benign tumors are classified as gangliocytomas or gangliogliomas. 13 A gangliocytoma is a tumor composed of mature ganglion cells positive for neuron-specific enolase and neurofilament protein. 13 A ganglioglioma is composed of astrocytic glial cells positive for glial fibrillary acidic protein as well as ganglion cells. 13 In the present case, the tumor exhibited a mixed population of glial fibrillary acidic protein–positive glial cells as well as neurofilament protein–positive and neuron-specific enolase–positive neuronal cells. Thus, we diagnosed the tumor as a ganglioglioma. These tumors are extremely rare in dogs; only a few cases have been reported. 19 Moreover, to our knowledge, no report has described ganglion cell tumors in the pituitary region of dogs.
Both pituitary gangliocytomas and pituitary gangliogliomas have been reported in humans. 7,8,12 Although these tumors are very rare, most cases involved concomitant pituitary adenomas. 7,8 An isolated pituitary hormone-secreting ganglion tumor causing an endocrine disorder has been reported less frequently. 8 In the present case, no concomitant corticotroph adenoma was identified, and the ganglion cells showed immunostaining for ACTH. This suggests that the ganglioglioma directly induced adrenal hypersecretion through ACTH secretion. Thus, we determined that the tumor must have been responsible for the hyperadrenocorticism. Because a typical corticotroph adenoma was not identified, classifying the present case as ectopic ACTH syndrome would be inappropriate. As the large tumor was presented within the entopic sella turcica, it is more suitable to recognize the ganglioglioma as a rare cause of PDH. In addition, the complete and prolonged remission of hypercorticism observed in the present case after surgery supported our diagnosis.
The precise histogenesis of the pituitary tumor in the present case remains unclear. In humans, the possible mechanisms for the development of pituitary gangliocytoma and ganglioglioma have been widely debated. 7,8,12 Briefly, in patients with mixed pituitary adenomas with ganglion tumors, the ganglion cells may be derived from the neurohypophysis, pituitary stalk, or hypothalamus. Furthermore, ganglion cells may produce some pituitary hormone–releasing hypothalamic hormones, thus stimulating abnormal proliferation of the anterior pituitary cells that results in endocrinological disease. 15 In the present case, however, the ganglion cells were immunohistochemically negative for CRH and arginine vasopressin, and no concomitant adenoma was identified. Therefore, this hypothesis is not suitable in the present case. Alternatively, the morphology of these tumors suggests that the ganglionic component originates from neuronal differentiation of adenoma cells in a process suggestive of transdifferentiation. This theory relies on the observation that ganglionic cells may express the same pituitary hormones as the associated adenoma. 7,8 If such differentiation occurred with glial differentiation in early tumorigenesis, and these components filled the majority of the tumor, it might explain the development of a pure ganglioglioma with ACTH hypersecretion. Meanwhile, the production of pro-opiomelanocortin—the precursor of ACTH—has been described in hypothalamic neurons. 4 If the ganglion cells were derived from such neurons, major differentiation might not be required to secrete ACTH. Finally, a common origin has been suggested for both neuronal and adenomatous components of uncommitted stem/progenitor cells in humans. 5 In addition, cells positive for stem/progenitor cell markers have been reported in the normal dog pituitary and in adenomas of dogs. 20 These findings might explain the origin of pituitary hormone–secreting neuronal/ganglionic cells from a progenitor cell. However, the histogenesis of a singular canine pituitary ganglion tumor is difficult to explain.
To our knowledge, this is the first case of hyperadrenocorticism caused by a pituitary ganglioglioma in a dog. The ganglioglioma appeared to be directly responsible for the hypersecretion of ACTH, and its removal by transsphenoidal surgery resulted in complete remission of the hyperadrenocorticism.
Footnotes
Acknowledgements
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.