
Abstract
The placenta is a vital organ providing the developing fetus with nutrient and gas exchange, thermoregulation, and waste elimination necessary for fetal development, as well as producing hormones to maintain pregnancy. It is hypothesized that fetal pig death in porcine reproductive and respiratory syndrome may be attributed to pathology of the maternal-fetal interface leading to premature placental separation. This study was designed to evaluate the chronologic progression of porcine reproductive and respiratory syndrome virus (PRRSV)–induced lesions at the maternal-fetal interface, with particular focus on placental separation in experimentally challenged third-trimester gilts. Fifteen gilts were inoculated with a virulent strain of PRRSV-2 on gestation day 86 ± 0.4. On multiple days postinoculation, 3 gilts along with 1 sham-inoculated control per time point were euthanized, and uterine and fetal placental tissues corresponding to each fetus were collected for histopathologic evaluation. The presence of any fetal lesion was 23 times more likely in compromised (meconium-stained and decomposed) compared with viable fetuses (P < .001). In PRRSV-infected gilts, endometritis was more severe than placentitis, and the severity of endometrial inflammation and vasculitis increased progressively from 2 to 14 days postinoculation. Neither placental vasculitis nor a chronologic progression in the severity of placental detachment was observed. Severe placental detachment was more frequently present in PRRSV-infected compared with noninfected samples and was most significantly associated with placental inflammation, compared with other uterine lesions, viral load, or termination day. The results of this study suggest that placental separation by itself is not sufficient to significantly compromise fetal viability in reproductive porcine reproductive and respiratory syndrome.
Keywords
In domestic animal species, fetal infection is reported to be one of the main mechanisms contributing to the pathogenesis of pregnancy failure of infectious causes. 24 However, because of the frequent absence of lethal lesions observed in the fetus, premature placental separation induced by in utero infection has been occasionally proposed as the main pathogenic mechanism of pregnancy failure in some infectious diseases. 30 For example, in horses, which have diffuse epitheliochorial placentation similar to porcine placenta, pathogens can often cause abortion by inducing inflammatory changes in the endometrium, production of cytokines, and damage to portions of the chorionic membrane leading to fetal hypoxia. 24 Abortions caused by equine arteritis virus are suggested to be specifically related to the inflammatory lesions in the uterus. Endometrial vasculitis with accompanying edema is reported to compromise blood flow, causing hypoxia of the placenta and decreased production of progesterone, leading to chorionic detachment and expulsion of equine arteritis virus–infected, and also noninfected, fetuses. 1 Similar to equine arteritis virus, another member of the Arterivirus family, porcine reproductive and respiratory syndrome virus (PRRSV), 27,28 is known to cause significant endometrial vasculitis and abortions in the third trimester of gestation. 12,18,21 However, the exact mechanism of fetal death is not fully understood.
Porcine reproductive and respiratory syndrome (PRRS) costs the North American swine industry $664 million annually, 5 with 45% of the losses due to reproductive failure. Understanding the pathophysiologic mechanism(s) responsible for fetal death related to PRRSV infection is an essential step in designing effective control measures against this costly disease. Historically, natural outbreaks and experimental studies of the reproductive form of PRRS confirmed relatively consistent maternal uterine pathology consisting of various severities of myometritis, endometritis, and placentitis accompanied with vasculitis, hemorrhage, and placental separations. Fetal lesions are infrequent, inconsistent, or completely absent. 13,22,24,26 It was initially proposed 13 that lesions associated with the placenta and uterus are responsible for the onset of abortions, suggesting disruption of the maternal-fetal barrier as the cause of fetal death. More recent studies also hypothesized that pathologic processes at the maternal-fetal interface (MFI) are responsible for the fetal death during PRRSV infection. 7,23
Our recent study of PRRSV-2 infection revealed lesions at the MFI (endometrium with adherent fetal placenta) at 21 days postinoculation (dpi) that appear to be incompatible with fetal survival , but because of the large numbers of autolyzed and decomposed fetuses, we failed to confirm the association between lesions at the MFI and fetal preservation status. 18 At 21 dpi, lesions in endometrium were advanced, and only the presence of lesions in the umbilical cords and fetal tissues were confirmed to be significantly associated with fetal death at that time point. The first goal of the present study was to assess temporal changes of PRRSV-induced specific pathology in fetuses between 2 and 14 dpi. The second goal was to determine group differences in the severity of pathologic lesions in the MFI (endometritis, placentitis, vasculitis, and placental separation) over time between PRRSV-infected and noninfected control gilts and between fetuses of different preservation statuses. These results led us to further investigate factors potentially associated with placental separation, including MFI lesion severity, postinoculation time point, fetal preservation status, and PRRS viral load in endometrium and fetal placenta.
Methods
Experimental Design
Twenty 6-month-old, purebred Landrace gilts from a PRRSV-naive farm were artificially inseminated using semen from Yorkshire boars. Fifteen pregnant gilts were intramuscularly and intranasally inoculated with PRRSV-2 (NLSV 97-7895; 1 × 10 5 50% tissue culture infective dose total dose) on gestation day 86 ± 0.4 and then euthanized at 2, 5, 8, 12, or 14 dpi (3 gilts per time point), along with one sham-inoculated control at each day postinoculation (5 total). An equivalent volume of minimum essential medium was similarly administered to all sham gilts on gestation day 86 ± 0.5. All gilts were vaccinated prebreeding against porcine parvovirus, porcine circovirus type 2, Leptospira interrogans (multivalent) and Erysipelothrix rhusiopathiae using commercial products. The study protocol adhered to principles established by the Canadian Council on Animal Care and was reviewed and approved by the Animal Research Ethics Board at the University of Saskatchewan (permit no. 20160023).
Gross Pathology
The gravid reproductive tract was removed intact and placed in a specially designed trough in a linear manner. The uterus was cut from left oviduct to right oviduct along the mesometrium (broad ligament) starting at the tip of each horn. Fetuses were numbered sequentially according to their position within each horn starting with “L1” and “R1,” closest to the ovary on the left and right sides, respectively. Sampling of the uterus with adherent fetal placenta (chorioallantois) was performed by cutting uterine wall (full thickness) into rectangular samples centered on the umbilical stump (∼20 cm long side) of each fetus. External fetal preservation status was assessed on the basis of previously published criteria 11 as follows: viable (VIA; normal, white to purple skin with visible hair and regular umbilical cord), meconium stained (MEC; skin covered with inspissated, brownish amniotic fluid and regular umbilical cord with edema), and decomposed (DEC; <50% of skin discolored, no blood in the umbilical cord or from axilla).
Histopathology
From the sample of MFI obtained at the necropsy, rectangular 3 × 1 cm, full-thickness samples adjacent to the umbilical stump of each fetus were fixed in 10% neutral buffered formalin solution and paraffin embedded for histopathologic evaluation. Histopathology of the MFI was assessed by a pathologist blinded to inoculation group, fetal preservation status, time points of infection, and PRRSV ribonucleic acid (RNA) concentration in the endometrium and fetal placenta. Severity of inflammatory lesions in endometrium and fetal placenta was separately assessed on the basis of a previously published grading scheme. 18 Briefly, the severity of the inflammatory cell infiltrate in endometrium (Figs. S1–S4) and chorioallantois (Figs. S5–S8) was graded as follows: normal = score 0 (no inflammatory cell infiltrate in the tissue), minimal = score 1 (<10% of tissue section contains inflammatory cell infiltrate), mild = score 2 (10%–25% of tissue section contains inflammatory cell infiltrate), moderate = score 3 (25%–50% of tissue section contains inflammatory cell infiltrate), and severe = score 4 (>50% of tissue section contains inflammatory cell infiltrate). The distribution of vasculitis, a PRRSV-specific lesion in MFI, was evaluated separately in the endometrium and fetal placenta on the basis of a previously published scoring scheme 18 : normal = score 0 (no blood vessels inflamed in the tissue section), focal = score 1 (<30% of inflamed blood vessels in the tissue section), multifocal = score 2 (30%–70% of inflamed blood vessels in the tissue), or diffuse = score 3 (>70% of inflamed blood vessels in the tissue sections inflamed) (Figs. S9–S12). Finally, separation of placenta from the uterus was assessed. To eliminate potential artificial placental separations that may have occurred during tissue processing, only placental detachments associated with inflammation in the endometrium and fetal placenta, and/or the presence of acute cellular swelling (degeneration) of trophoblastic epithelium, and/or the presence of necrotic debris was used for evaluation because they require time to develop and therefore were considered to have occurred antemortem. The severity of these detachments was evaluated using a novel grading scheme that reflected the percentage of maternal-fetal interdigitation area affected by this pathologic process. Therefore, the severity of separation was scored as follows: normal = score 0 (no detachment of placenta along the interdigitation area), mild = score 1 (<25% of interdigitation area affected by detachment of placenta), moderate = score 2 (25%–50% of interdigitation area affected by detachment of placenta), and severe = score 3 (>50% of interdigitation area affected by detachment of placenta) (Figs. S13–S16).
Quantification of PRRSV RNA Concentration
Samples of MFI corresponding to each fetus were placed in weigh boats (endometrium and placenta facing up, myometrium facing down) on ice for up to 45 minutes. After gently washing, they were placed with myometrium side down on absorbent tissue, where the fetal placenta was manually separated from endometrium using sterile forceps. RNA extraction from placental and endometrial samples and quantitative polymerase chain reaction (PCR) targeting PRRSV NVSL 97-7895 was performed as previously described in detail. 11 Briefly, RNA was extracted from 30 mg of tissue using the RNeasy MiniKit (QIAGEN, Toronto, ON, Canada) performed on the Qiacube Robotic workstation, and RNA concentration and quality (A260/A280 ∼ 2) were assessed using 2 μL of each sample with a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA). Primers and probe were designed to target a highly conserved region at the C-terminal end of ORF7 of NVSL-97-7895. Reactions (20 μL) contained 2 μL RNA extract, 10 μL Mastermix (Bio-Rad iTaq Univeral Probes 1-Step Kit; Bio-Rad Laboratories, Mississauga, ON, Canada), 0.5 μL reverse-transcriptase/RNase block enzyme Supermix (iScript Reverse transcriptase; Bio-Rad), 0.5 μL of 10 μM PRRS-2F primer 5′-TAA TGG GCT GGC ATT CCT-3′, 0.5 μL of 10 μM PRRS-1 R primer 5′-ACA CGG TCG CCC TAA TTG-3′, 0.5 μL of 10 μM PRRS-P1 probe 5′-HEX-TGT GGT GAA TGG CAC TGA TTG RCA-BHQ2-3′ (reported initially by Kleiboeker et al 8 ), and 6 μL RNAse-free water. Each PCR plate contained 35 samples in duplicate, extraction controls, positive control (known positive tissue homogenate), and a quantitative PCR nontemplate control. A dilution series (10 7 –101 copies/μL) of HindIII linearized plasmid, pCR2.1TOPO-NVSL, containing a 446-bp sequence of ORF7 was used as a standard curve, run in triplicate on each plate. The PCR protocol was reverse transcription for 30 minutes at 50°C followed by 10 minutes at 95°C, 40 cycles of denaturation for 30 seconds at 95°C, and annealing/extension for 30 seconds at 59°C. Individual samples were rerun if the Cq standard deviation between the duplicates was >1.5 or if 1 of the 2 duplicates had no Ct. Results were reported as log10 target RNA concentration per milligram of tissue. The limits of quantification were defined by the least and most concentrated standards. Samples were recorded as negative if target RNA was not detected or detected but not quantifiable if target RNA was detected but the concentration was below the least concentrated standard. All PCR plates contained appropriate nontemplate and extraction controls and a positive control sample (serum or tissue as appropriate) to track interplate variation. Across all plates, the PCR reaction efficiency averaged 99.85 ± 4.94%.
Statistical Analyses
All statistical analyses were performed using Stata 14 (StataCorp, College Station, TX). Descriptive statistics were used to summarize the chronologic progression of fetal lesions over the 5 termination days. The frequency of fetal lesions in VIA compared with compromised (MEC + DEC) fetuses was assessed using a Fisher exact test. MEC and DEC fetuses were combined into a single group (compromised) because there was an insufficient number in each group. Group differences (PRRSV inoculated vs noninoculated) in the severity of each lesion (endometritis, placentitis, vasculitis in the endometrium and fetal placenta, placental separation) were evaluated using a 2-sided Fisher exact test, first by assessing differences across all 5 termination days together and, if significant, by subsequently looking at each individual day. To validate these results, differences in median lesion severity scores between PRRSV inoculated and noninoculated were assessed at each termination day using a Mann-Whitney U test. To determine if lesion severity progressed over time in the PRRSV-inoculated group, median lesion severity scores were compared across termination day using a Kruskal-Wallis test, followed by a post hoc Dunn test (with Sidak multiple comparison adjustment). Finally, the association between the placental separation severity score in the PRRSV-infected samples and PRRS viral load in the endometrium and fetal placenta, severity of endometritis, placentitis and vasculitis at MFI, termination day, and fetal preservation status was analyzed using single-level proportional odds model. For the purpose of these analyses, the values for PRRSV RNA concentration in the endometrium and placenta and placental separation severity scores were categorized on the basis of the natural break points in data. Endometrial viral load categories were 0 if PRRSV RNA concentration was <3 log10 copies/mg, 1 if ≥3 and ≤5.75, and 2 if >5.75. Placental viral load categories were 0 if PRRSV RNA concentration was <2 log10 copies/mg, 1 if ≥2 and ≤5.5, and 2 if > 5.5. For placental separation, because of relatively small numbers of samples with moderate and severe scores compared with normal and mild severity, the moderate and severe severity categories were combined.
Results
The average litter size over 20 inoculated and control gilts was 13.8 (range, 8–22). No gilts aborted following inoculation, and the study population included 205 and 71 fetuses from PRRSV-inoculated and control gilts, respectively (276 fetuses total). All control fetuses were VIA, except for 1 MEC. Just over 90% of fetuses from PRRSV-infected gilts were VIA (185 of 205), with the remainder being MEC (13 of 205 [6.3%]) or DEC (7 of 205 [3.4%]).
Fetal Histopathology
The prevalence of fetal histologic lesions by termination day are summarized in Table S1. The presence of any fetal lesion was 23 times (95% confidence interval, 8.5–62.5) more likely in compromised (MEC + DEC; 13 of 22 [59.1%]) compared with VIA (15 of 239 [5.9%]) fetuses (P < .001). Pathologic changes in fetal tissues observed were minimal to mild in severity and not specific. In total, 276 fetal brains, lungs, hearts, and livers, 228 thymuses, and 219 mesenteric lymph nodes were examined. The first pathologic change in fetal organs was observed 5 dpi in the thalamus and consisted of mild focal areas of gliosis (samples of diencephalon were systematically collected in this study to assess potential neuronal necrosis, particularly in the hypoxia-sensitive hippocampus). 31 Similar areas of mild gliosis were also observed in the cortex of diencephalon 8, 12, and 14 dpi. This change was not observed in negative control fetuses. Also, no neuronal necrosis was observed in the PRRSV-infected or negative control samples. Lesions in heart, previously reported to be consistent fetal lesions of PRRS, 22 were first observed in this study 8 dpi and consisted of small aggregates of lymphocytes between cardiomyocytes (mild focal lymphocytic myocarditis), but there was no dramatic change in the prevalence or severity of this lesion over time. No necrosis of cardiomyocytes was seen in this study, including in the negative control samples. The only pathologic change observed in the lung was the occasional presence of small to moderate amounts of meconium in the airways, which was seen starting from 8 dpi and only in MEC and DEC fetuses. Meconium in the bronchioles was also seen in 1 MEC negative control fetus. One PRRSV-infected fetus exhibited a mild increase in lymphocytolysis in the cortex of the thymus 12 dpi, but no thymic atrophy was observed in the whole study population. Finally, 14 dpi, grossly enlarged mesenteric lymph nodes were observed in 5 fetuses at necropsy that microscopically corresponded to mild to moderate follicular hyperplasia (reactive).
Histopathology of the MFI
Results of histopathologic lesions at the MFI are summarized in Table S2. The earliest lesion in the endometrium of PRRSV-infected gilts was focal vasculitis observed 2 dpi affecting medium-size venules in the endometrium (Fig. 1). Focal vasculitis (<30% blood vessels affected) was characterized by accumulation of small numbers of lymphocytes and histiocytes under the endothelial lining without associated damage. By 8 dpi, more than half of the PRRSV-infected samples exhibited multifocal (30%–70% blood vessels) or diffuse (>70% blood vessels) endometrial vasculitis. On 12 dpi, all PRRSV-infected samples exhibited varying degrees of vasculitis in the endometrium, with the majority of samples having a diffuse pattern of distribution (Fig. 2). The severity of vasculitis increased across days, progressively affecting medium-size arterioles and occasionally larger veins, but the predominant inflammatory cell types of vasculitis, lymphocytes, and histiocytes remained the same. The endothelial cell lining remained intact in endometrial tissue samples on all days postinoculation. No vasculitis was observed in PRRSV-infected fetal placental samples or in the endometrium or placenta of noninfected control samples.
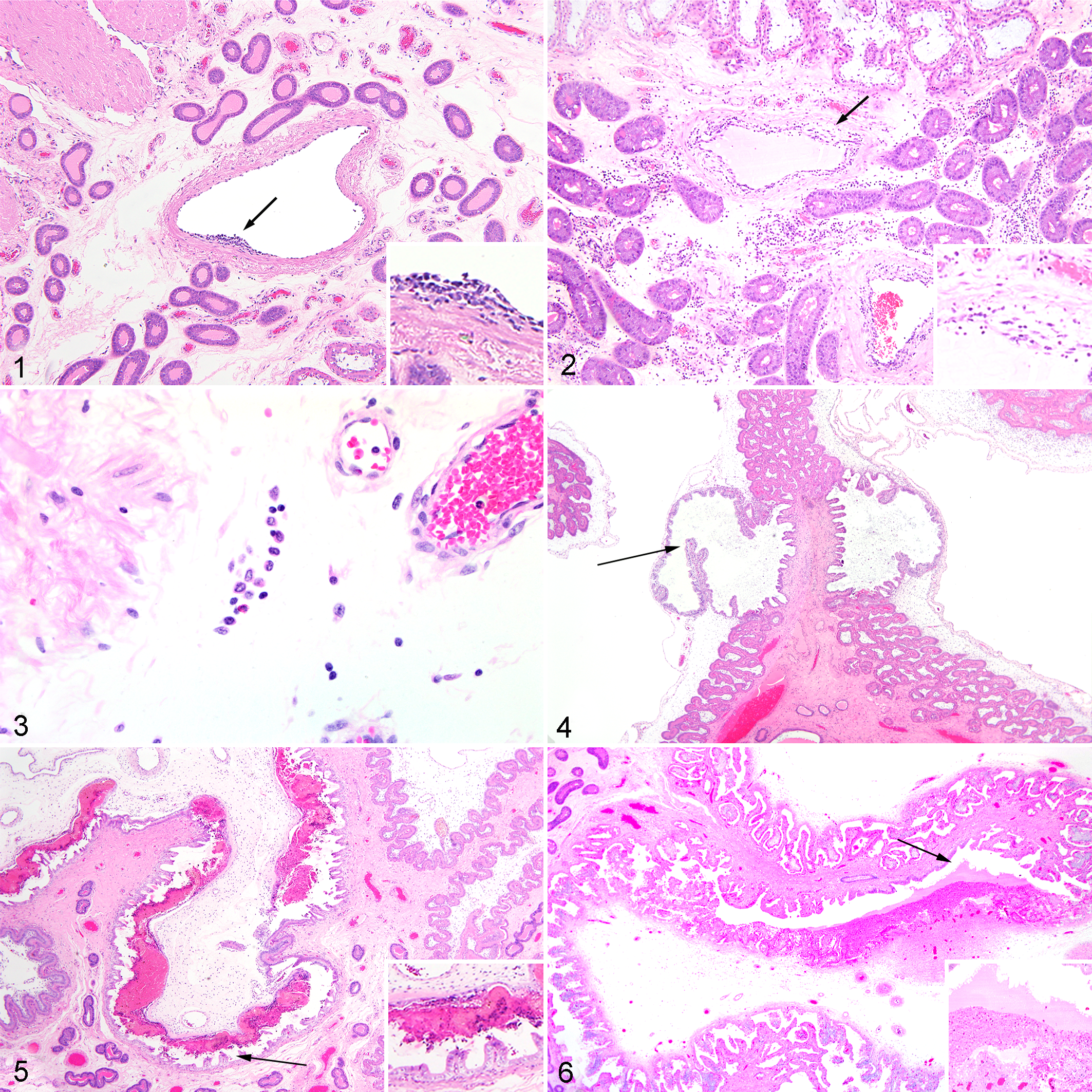
In PRRSV-infected animals, severe endometritis was observed for the first time 8 dpi, and by 14 dpi, more than half of samples were affected. However, placentitis in PRRSV-infected samples differed from endometritis in its severity (mostly milder, rarely moderate or severe) and by type of inflammatory cells involved. Severe placentitis was seen in only a single sample at 12 dpi. Although endometritis was characterized by large numbers of lymphocytes, small numbers of histiocytes, and rare plasma cells, placentitis was composed exclusively of varying numbers of plump vacuolated macrophages residing along the placental mesenchyme in row-like formation in the area between MFI and placental vasculature (Fig. 3). PRRSV-negative control animals had either normal endometrium or minimal endometritis, except for single samples at 2, 12, and 14 dpi that scored mild, mild, and moderate, respectively. As for placentitis in control animals, all samples were normal or minimal, except for a single 14-dpi sample that scored mild.
Placental separation was observed only microscopically (not grossly) and characterized mainly by moderate to severe inflammation in the endometrium and fetal placenta, marked acute vacuolar degeneration of detached trophoblastic epithelium, mild to moderate focal areas of necrosis of uterine epithelium, and varying amounts of necrotic debris in the detachment gaps. Mild separation was commonly observed in noninfected control gilts. Moderate and severe placental separation were each observed in 1 noninfected fetus, both VIA. The only noninoculated (control) MEC fetus showed no evidence of placental separation. In the PRRSV-infected MFI samples, frequent areas of mild (Fig. 4) and moderate (Fig. 5) microscopic placental separation were both seen from 2 to 8 dpi. Severe placental separation (Fig. 6) was observed 12 and 14 dpi only.
Group Differences in Microscopic Lesions at the MFI
Potential differences in lesion severity in MFI samples from PRRSV-inoculated and noninoculated gilts were assessed using 2 methods. In the first assessment method using contingency tables, Fisher exact tests revealed that the endometrial vasculitis, endometritis, and placentitis severity scores differed significantly in PRRSV-inoculated compared with negative control samples (P < .001 for all) when all days were considered together. Further analyses of group differences in these lesion scores at each termination day revealed that endometritis severity (Fig. 7) differed on all 5 termination days, whereas placentitis severity (Fig. 8) and vasculitis severity (Fig. 9) differed at 5 to 14 dpi but not 2 dpi, indicating that endometrial vasculitis and placentitis require more time to develop. Although placental separation severity scores (Fig. 10) also differed between PRRSV-inoculated and noninoculated samples when all days were considered together (P = .037), the group differences were driven mainly by a significant difference in severity at 5 dpi.
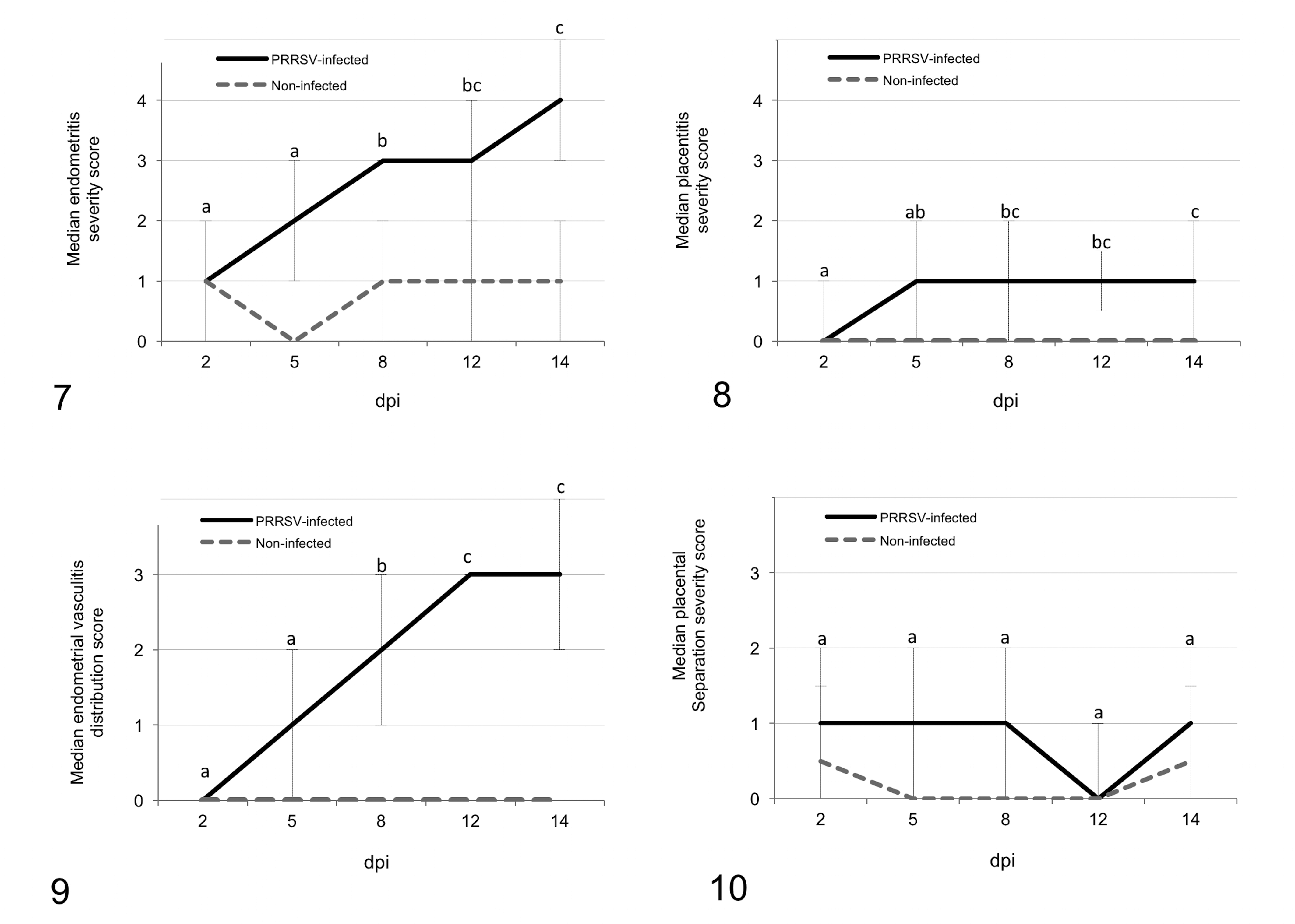
The second method of assessment involved the comparison of median lesion scores by group (Mann-Whitney U test), initially across all termination days, then individually by day. Differences in median rank lesion severity scores between PRRSV-inoculated and noninoculated samples were identical to those using the contingency method.
Progression of Microscopic Lesions at the MFI Over Time
These analyses included samples from PRRSV-inoculated gilts only and were based on the hypothesis that lesion severity at the MFI would increase over time, potentially leading to more severe placental separation. Spatiotemporal changes in the severity of vasculitis, endometritis, placentitis, and placental separation are shown in heat maps in which darker colors represent increased severity (Figs. S17–S20). On the basis of a Dunn test, the median severity ranks for endometritis, placentitis, and endometrial vasculitis increased over time (P < .001 for all) (Figs. 7–9). Surprisingly, placental separation (Fig. 10) did not differ across day of termination, when all PRRSV-inoculated fetuses were considered (P = .85), or when only considering the viable fetuses (P = .56).
Factors Associated With Placental Separation
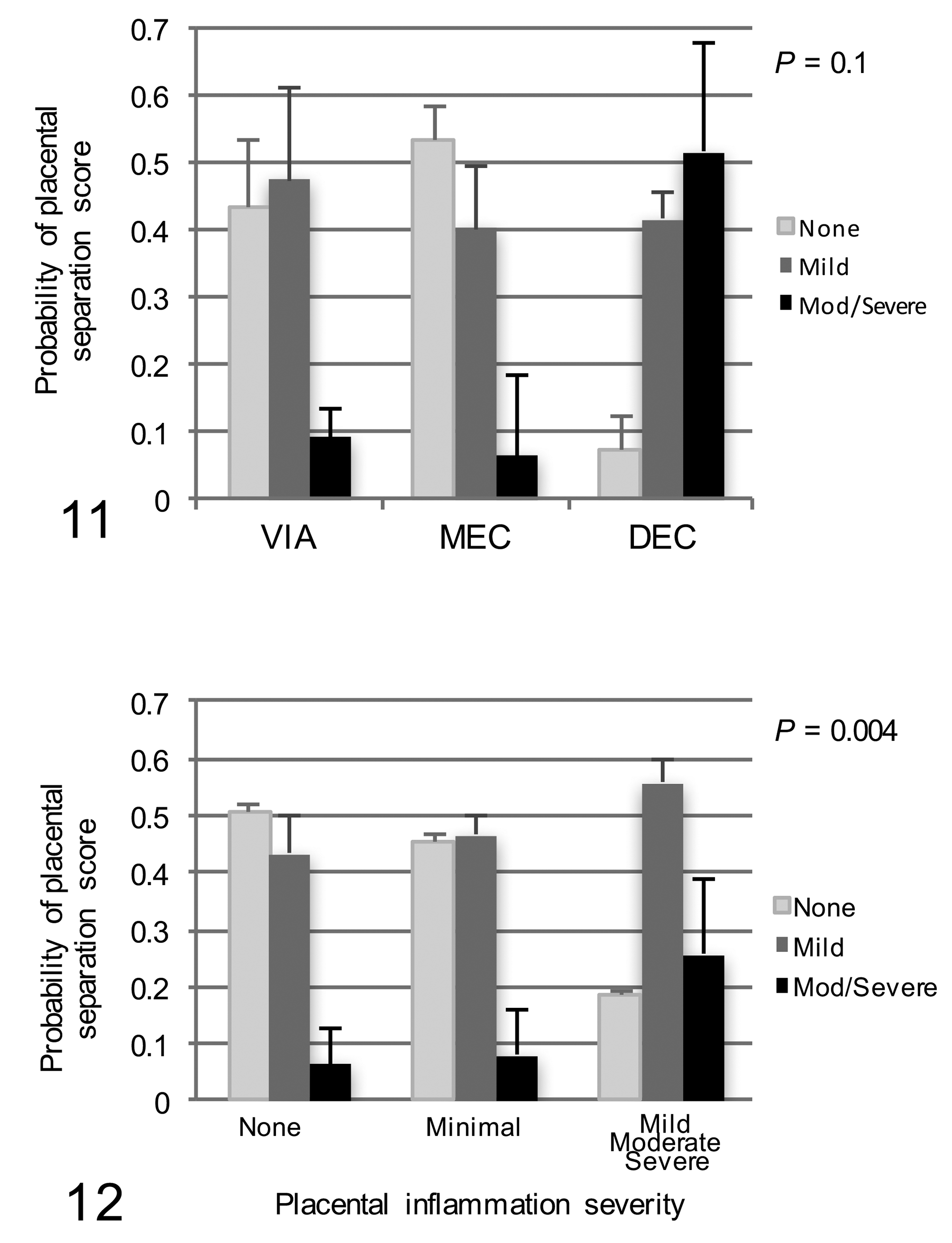
Having found wide spatiotemporal variation in placental separation scores (Fig. S20) but no progression in severity over time in the PRRSV-inoculated group, together with overall group differences in severity mainly driven by a single day (5 dpi), a proportional odds model was used to better understand factors potentially associated with placental separation severity. Although the study population was naturally clustered by gilt, a 2-level hierarchical model was not required. Of 7 factors unconditionally evaluated, only placental inflammation severity, vasculitis distribution, and fetal preservation were unconditionally associated with placental separation severity (P < .20) and thus included in the full model. Importantly, termination day, endometritis severity, and viral load in endometrium and placenta were not associated with placental separation score. After backward stepwise elimination, placentitis severity remained the most significant factor associated with placental separation (P < .004), whereas fetal preservation score only trended toward significance (P = .10). Post hoc pairwise analysis revealed that placental separation severity was associated only with fetal death; the probability of a DEC fetus having moderate or severe placental separation was .51 ± .16, compared with .06 ± .04 for MEC and .09 ± .05 for VIA fetuses (Fig. 11). The relationship of placental separation severity to placentitis is shown in Fig. 12. The probability of severe or moderate placental separation was .26 ± .13 if placentitis severity was mild to severe, compared with less than .08 if placentitis was minimal or none.
Discussion
The aim of this study was to evaluate the chronologic progression of lesions occurring in the MFI and in adjacent fetuses in gilts experimentally challenged with PRRSV-2 and to assess factors associated with placental separation. The study confirmed the chronologic progression in the severity of inflammatory lesions in the endometrium, fetal placenta, and endometrial vasculature of PRRSV-inoculated gilts but failed to confirm the presence of inflammation in placental vasculature as well as any progression in the severity of placental separation over time.
PRRSV-2 infected uterine tissues by 2 dpi, specifically small to medium-caliber venous blood vessels, without damaging endothelial cells. At 2 dpi, the lumen of many endometrial blood vessels contained increased numbers of mononuclear leukocytes with unilobar nuclei (presumed monocytes) in close proximity to endothelium and occasionally clearly adhered to it. The lack of endothelial cell damage and hypertrophy of endothelial cells (reactive) with adherent monocytes are indicative of initiation of the leukocyte adhesion cascade as a mechanism for viral entry subintimally into blood vessel walls and further into the endometrial stroma. Therefore, the vasculitis and endometritis are likely the consequence of an immediate (innate) immune response, involving endometrial histiocytes, natural killer cells, and lymphocytes, to the entry of viral-infected monocytes into the subintima forming small foci of vasculitis. The stimulus for expression of adhesion receptors (selectins) of the endothelium or activation of endothelium for the leukocyte adhesion cascade is unclear, because PRRSV infection does not induce significant secretion of cytokines such as interleukin-1 and tumor necrosis factor–α in gilt blood. 10,14 More likely, the stimulus for entry is provided by a chemoattractant from the endometrium, which needs to be explored more in the future.
An interesting finding in the present study was the absence of vasculitis in all chorioallantoic and umbilical cord blood vessels at all time points between 2 and 14 dpi. By contrast, PRRSV-induced vasculitis of endometrial vasculature was commonly observed in this study as well as in previous studies. 13,18,26 PRRSV-induced vasculitis in fetal tissues has been reported only in umbilical cord arteries. 13 Together, this finding suggests substantial differences in the response to PRRSV infection between the maternal and fetal reproductive compartments. 9,23,29 In concert with the absence of placental vasculitis, placentitis was less severe than endometritis in PRRSV-infected samples. The placentitis was composed primarily of a homogenous population of plump, vacuolated macrophages previously confirmed to be mostly CD163 positive. 20 The meager placental inflammatory response suggests that PRRSV may induce a degree of fetal immunosuppression that impedes the infiltration of inflammatory cells to prevent placental vasculitis. Evidence for this is a significant decrease in the number of CD163-positive macrophages, the main target cell population for PRRSV replication, which is observed during experimental PRRSV infection 21 dpi, 20 as well as our understanding that the fetal thymus is likely the primary site of virus replication. 23 Alternatively, the distinctive inflammatory responses in endometrium and fetal placenta, and the lack of fetal placental vasculitis could be the result of differing chemokine and cytokine environments and gradients between the uterus and fetal placenta (proinflammatory vs inflammatory) 2 or reflect the limited capacity of the developing fetal immune system to react following PRRSV challenge. 25 Finally, the lack of vasculitis in the fetal placenta could be due merely to differences in the specific receptors for virus attachment on endothelial cells in uterine and fetal placental blood vessels. Porcine endometrial endothelial cells expressing sialoadhesin (CD169) and CD151 but not CD163 support PRRSV growth in vitro. 3 Whether in vivo endometrial endothelial cells, in contrast to placental endothelial cells, are permissive to viral infection or only serve for trafficking the virus needs more clarification.
The pathogenesis of fetal death during PRRSV infection is still a matter of debate. On the basis of our current knowledge, 4,9 events in the MFI and fetus appear to contribute to the fetal demise, but the relative importance of each is not known. On one hand, the presence of viral RNA in fetal blood, lymphatic organs, lung, and heart, 23 the clustering of virus-infected and dead fetuses in litters, 9 and the upregulation of genes in fetal thymus indicative of inflammatory responses and disease progression 29 provide evidence that the pathogenesis of fetal death may be the result of transplacental infection of fetal tissues and subsequent events occurring in fetuses. This and a previous study 18 have also demonstrated that although fetal histologic lesions are rare, they are strongly associated with the likelihood of fetal death. On the other hand, the rarity to complete absence of lesions in fetal organs has been used by others 12,13,16,18,22 to emphasize the importance of events occurring at the MFI. Specifically, the presence of significant pathology in the endometrium, 18 increased apoptosis at the implantation sites, 7,19 and the association of fetal death with PRRSV concentration in the implantation sites 9 all suggest that disruption of the MFI, placental separation, and subsequent placental dysfunction may be the main mechanisms of fetal death. 6,7,23
Separation of trophoblastic epithelial cells from the maternal epithelium related to lack of oxygen supply and/or nutrition (lack of histotroph and hematotroph) inevitably leads over time to hypoperfusion, cell hypoxia, vacuolar swelling (degeneration), cell death (necrosis), and phagocyte clearing (inflammation). Until now, no studies have chronologically assessed the severity of placental separation during the course of PRRSV infection in the third trimester of gestation. To avoid the introduction of artificial separations due to sample handling and/or processing, sections of uterus with adherent fetal placenta were initially evaluated for the presence of pathologic processes, and only those areas of placental separation that had concurrent necrosis, degeneration, and inflammation indicative of antemortem change were subjected to further semiquantitative assessment. However, the main limitation of our study was an inability to distinguish placental separations on the basis of their specific pathogenetic mechanisms, such as those that were virally induced (cytolytic effect, cytokine bystander effect, inflammation) or physiologic (local cytokine disbalance, altered interactions between uterine epithelium and fetal trophoblast). Because such classification of placental separations would be exceptionally challenging in multiparous species, we assessed the severity of placental separation in samples collected from PRRSV-infected compared with negative control gilts that were processed simultaneously and under similar conditions. This study confirmed that the severity of placental separation was significantly increased in MFI samples of PRRSV-infected compared with noninfected control gilts. Furthermore, the severity of placentitis was the most significant factor associated with severity of placental separation at the MFI. This is an important finding, particularly in light of knowledge that PRRSV induces cell death (apoptosis) of trophoblastic cells and uterine epithelial cells and is postulated to be a potential mechanism of fetal death. 7,19 With this as background, we hypothesized a priori that more severe placental separation should be evident in MEC fetuses compared with VIA fetuses, because meconium staining is indicative of PRRSV-associated fetal compromise 11 and disease progression. Unexpectedly, the present study not only confirmed the absence of significant placental separations during the 14-day course of PRRSV infection but also revealed no increase in severity associated with meconium staining.
Increased severity of placental separation was observed in decomposed PRRSV-infected fetuses, but it is still unclear whether placental separation is a cause or an effect of fetal death. On one hand, placental separation observed in decomposed fetuses may be due to postmortem autolysis of placental mesenchyme. On the other hand, the absence of severe placental separation associated with meconium staining indicates that separation by itself is not sufficient to compromise fetal viability and that PRRS-related fetal compromise is likely associated with other pathophysiologic events occurring in fetal tissues, in the MFI, or both. It is important to note that in the present and our previous studies, 11 meconium staining was rarely observed in fetuses of noninoculated control gilts at termination. In our view, this provides evidence that several other potential mechanisms of fetal stress that should be equally prevalent in fetuses of control and PRRSV-inoculated gilts, including prolonged or severe intrauterine hypoxia, umbilical cord torsion, and maternal infection that compromise fetal blood supply, 15,17 are also not primary mechanisms of fetal death following PRRSV infection. The fact that TWIST1, a transcription factor involved in fetal growth, angiogenesis, placental development, and implantation, is inhibited following PRRSV-2 infection 29 suggests that PRRSV infection adversely affects other aspects of placental function that may not directly involve detachment. Admittedly, it is prudent to exercise caution when interpreting the results of a single experiment, and an important limitation of the present study is that histopathology and viral load were assessed in only 1 subsample collected from the umbilical stump region of each fetus, rather than from multiple areas spread across the entire organ. Nevertheless, the lack of association of placental separation severity with PRRS viral load in endometrium and fetal placenta, severity of endometritis, and distribution of endometrial vasculitis, along with the strong positive relationship between fetal lesions and likelihood of fetal death, also support our conclusion that placental separation in reproductive PRRS is not the sole factor in the pathogenesis of PRRSV-related fetal death. Additional studies, including chronologic investigations across a range of gestational periods in third trimester and beyond 14 dpi, using more sophisticated methods of assessing placental separation severity, and across a broader cross-section of MFI of each fetus are required to further validate our findings.
Conclusions
This study contributes to a better understanding of the pathophysiology of reproductive PRRS. First, it confirms a lack of fetal placental vasculitis despite a progression of inflammatory lesions in the endometrium, fetal placenta, and endometrial blood vessels. Second, it confirms that placental separations were most strongly associated with placental inflammation. However, the severity of placental separation did not progress during the course of PRRSV infection and was not associated with meconium staining of fetuses. Together with the fact that placental separation was most severe in decomposed fetuses suggests that separation by itself is not sufficient to significantly compromise fetal viability in reproductive PRRS.
Supplemental Material
Supplemental Material, DS1_VET_10.1177_0300985818765067 - Histologic Changes Associated With Placental Separation in Gilts Infected with Porcine Reproductive and Respiratory Syndrome Virus
Supplemental Material, DS1_VET_10.1177_0300985818765067 for Histologic Changes Associated With Placental Separation in Gilts Infected with Porcine Reproductive and Respiratory Syndrome Virus by Predrag Novakovic, Susan E. Detmer, Muhammad Suleman, Carol M. Malgarin, Daniel J. MacPhee, and John C. S. Harding in Veterinary Pathology
Footnotes
Acknowledgments
We express special acknowledgment to numerous personnel (technicians, graduate students, and summer students) helping in the preparation and implementation of the pilot study for Pregnant Gilt Model 2. We also acknowledge Larhonda Sobchishin of the Department of Veterinary Pathology for her assistance preparing the images.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was supported by Genome Canada, Genome Prairie (Saskatchewan Ministry of Agriculture) and Genome Alberta.
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.