
Abstract
Canine Lafora disease (LD) is an autosomal recessive genetic disorder causing nonfatal structural epilepsy, mainly affecting miniature wirehaired dachshunds. Repeat expansion in the EPM2B gene causes a functional impairment of the ubiquitin ligase malin which regulates glycogen metabolism. Abnormally structured glycogen accumulates and develop polyglucosan bodies predominantly in the central nervous system. The authors performed a comprehensive clinical, genetic, and pathological study of 4 LD cases affecting miniature wirehaired dachshund dogs with EPM2B repeat expansions, with systemic distribution of polyglucosan bodies and accumulation of laforin and other functionally associated proteins in the polyglucosan bodies. Myoclonic seizures first appeared at 7–9 years of age, and the dogs died at 14–16 years of age. Immunohistochemistry for calbindin revealed that the polyglucosan bodies were located in the cell bodies and dendritic processes of Purkinje cells. Polyglucosan bodies were also positive for laforin, hsp70, α/β-synuclein, ubiquitin, LC3, and p62. Laforin-positive polyglucosan bodies were located in neurofilament-positive neurons but not in GFAP-positive astrocytes. In nonneural tissues, periodic acid-Schiff (PAS)-positive polyglucosan bodies were observed in the heart, skeletal muscle, liver, apocrine sweat gland, and smooth muscle layer of the urinary bladder. In the skeletal muscle, polyglucosan bodies were observed only in type 1 fibers and not in type 2 fibers. The results indicate that although the repeat expansion of the EPM2B gene is specific to dogs, the immunohistochemical properties of polyglucosan body in canine LD are comparable to human LD. However, important phenotypic variations exist between the 2 species including the affected skeletal muscle fiber type.
Keywords
Lafora disease (LD) is an autosomal recessive disorder that causes progressive structural epilepsy. 3,6 The disease is histopathologically characterized by the presence of polyglucosan bodies (Lafora bodies), predominantly in the central nervous system. In humans, LD is a fatal disease that has an onset in the teenage years and causes death within 10 years after clinical manifestation. Adult polyglucosan body disease, another genetic disease with development of polyglucosan bodies in the nervous system, is caused by glycogen branching enzyme deficiency and clinically characterized by paresthesia and bladder dysfunction beginning in the 5th decade of life. 19 More than 90% of human LD cases are caused by a mutation in the EPM2A or EPM2B (NHLRC1) gene, resulting in aberration of the proteins they encode, laforin and malin, respectively. Laforin has a carbohydrate-binding module (CBM) and a dual specificity phosphatase (DSP) domain, and dephosphorylates glycogen. Malin is a ubiquitin ligase that has been shown to ubiquitinate laforin and promote its degradation. Moreover, malin forms a complex with laforin and ubiquitinates enzymes that are involved in glycogen synthesis in a laforin-dependent manner. Therefore, dysfunction of laforin or malin results in the accumulation of glycogen and glycosaminoglycans, which then causes the formation of polyglucosan bodies. Laforin and malin are also involved in protein folding and protein degradation in association with the chaperone proteins, the ubiquitin proteasome system, and autophagy. 8 Laforin positively regulates autophagy by increasing the expression of LC3, a protein involved in the formation of autophagosomes. 1 Moreover, since both laforin- and malin-deficient mice show a defect in autophagy, it is considered that lack of laforin-malin complex is responsible for the autophagy defect. 5,9 Dysfunction of these pathways plays an important role in neuronal damage and dysfunction and progression of LD.
In animals, LD has been reported in the dog, cat, cow, and fennec fox. 14,15,28 However, a genetic abnormality has been identified only in canine LD, in association with a species-specific repeat expansion in the EPM2B gene. 17 Miniature wirehaired dachshunds are the breed most commonly affected by the genetic defects. In a large study on miniature wirehaired dachshunds in the UK, nearly half of the dogs had at least 1 copy, and approximately 13% had 2 copies of the aberrant gene. 24 Recently, an owner-based retrospective study had demonstrated clinical phenotypes at early and late stage of the disease in dogs homozygous for the expanded EPM2B gene. 29 LD cases with EPM2B repeat expansion have also been reported in the basset hound and beagle. 13,17 Expression of malin is significantly decreased in affected dogs; 17 however, other proteins that are associated with the pathology of LD (ie, laforin and molecular chaperone-related, ubiquitin proteasome system-related and autophagy-related proteins) have not been examined in canine LD cases with EPM2B repeat expansion.
The diagnosis of LD is based on functional (clinical signs), genotypic (gene mutation), and phenotypic (histopathological findings) examinations. In humans, aside from genetic examination, detection of polyglucosan body in eccrine duct cells and peripheral nerve of skin biopsy is the least invasive and best established method for confirming LD by histopathology. 20 Antemortem diagnosis is also important in dogs because most canine LD cases present myoclonic episodes after sexual maturity, and affected dogs may live as long as nonaffected dogs. Thus, the genetic defects are difficult to eradicate from certain breeds.
In the present study, we performed a comprehensive clinical, genetic, and pathological study of canine LD cases. In addition, novel findings on the accumulation of laforin and other functionally associated proteins are discussed. The aim of this study was to demonstrate the systemic distribution of polyglucosan bodies and to assess the involvement of the chaperone proteins, ubiquitin proteasome system, and autophagy in canine LD with EPM2B repeat expansion.
Materials and Methods
Animals
Four miniature wirehaired dachshund dogs with signs of progressive myoclonic seizure were examined (Table 1). All dogs were tested for EPM2B repeat expansion after clinical onset. Magnetic resonance imaging was performed within a year after clinical manifestation. One dog (case 1) was used for breeding prior to disease onset. Myotonic seizure was controlled by the administration of antiepileptic drugs. Necropsies were performed at the University of Tokyo. Brains from 3 miniature dachshunds (14, 15, and 16 years old) without LD were also examined for histopathology and immunohistochemistry (Table 1).
Cases and Controls Examined in the Present Study.
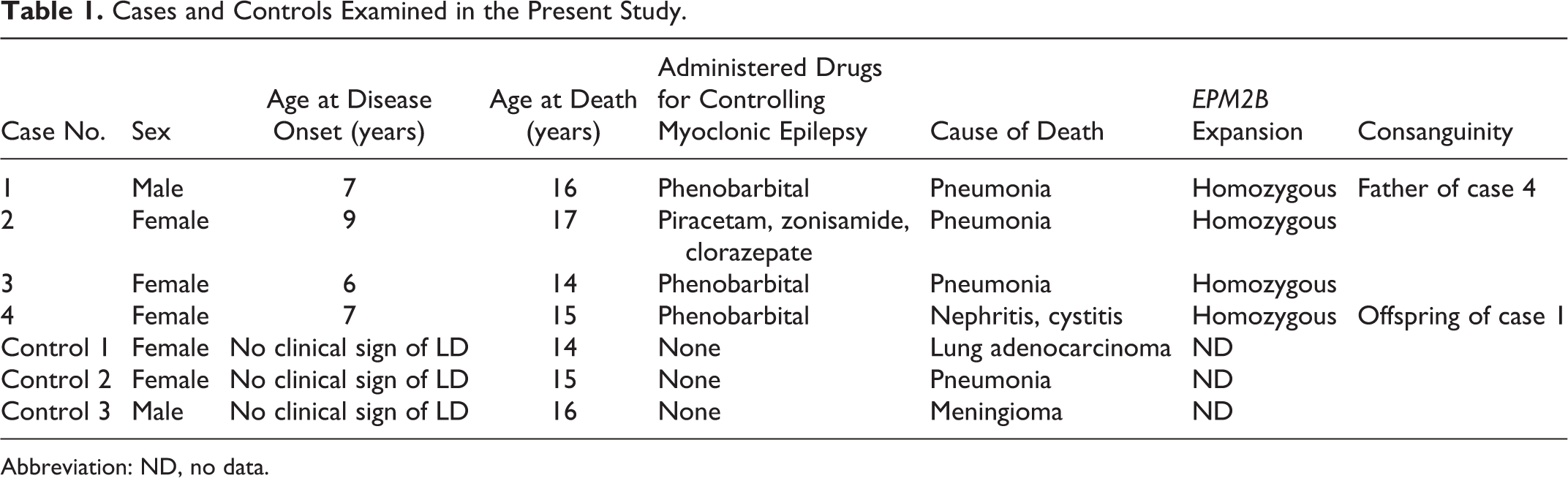
Abbreviation: ND, no data.
EPM2B Gene PCR Analysis
Total DNA was extracted from peripheral blood samples from the dogs and polymerase chain reaction (PCR) assay was performed as previously described. 17 EPM2B repeat expansion was determined by comparing the electrophoretic patterns of the PCR products. Total DNA samples from nonaffected (ie, no neurological signs) cases that were homozygous for the wild-type EPM2B gene and heterozygous for the repeat expanded EPM2B gene were used as controls.
Histopathology
Tissues were collected at necropsy, fixed in 10% neutral-buffered formalin solution, and then embedded in paraffin using a standard protocol. Tissues of the cerebellum, eye, and liver were fixed in methanol-Carnoy (Methacarn) solution. Four-micrometer serial sections were cut and stained with hematoxylin and eosin, periodic acid-Schiff (PAS), and Alcian blue (pH 2.5). The following tissues were examined for PAS-positive polyglucosan bodies: cerebrum, cerebellum, diencephalon, pons, medulla oblongata, spinal cord, retina, trigeminal nerve, sensory ganglion (cervical dorsal root ganglion), choroid plexus, liver, apocrine sweat gland, heart, skeletal muscle, stomach, small intestine, urinary bladder, and spleen. The trigeminal nerve and skeletal muscle were not examined in case 3. The skeletal muscles included the longissimus and triceps brachii in case 1; the lateral rectus, longissimus, and triceps brachii in case 2; and the diaphragm, esophagus, longissimus, oblique abdominal, and quadriceps femoris in case 4.
Microscopic images of the transverse sections of cerebral cortex were captured under x200 magnification. Horizontal images of the cortex in 3 areas (vermis and both hemispheres) were taken in each case. The number of Purkinje cells per view were counted manually, and the thicknesses of cerebral cortex were measured using Adobe Photoshop CS6 software (Adobe Systems, San Jose, CA) in each area. Comparisons of the means between LD (n = 4) and non-LD (n = 3) cases were performed by Student’s t-test. Differences with a P value < .01 were considered significant.
Immunohistochemistry
After deparaffinization and antigen retrieval, the sections were treated with 3% hydrogen peroxide in methanol at room temperature for 5 minutes, and then incubated with 8% skimmed milk in Tris-buffered saline at 37°C for 40 minutes to block nonspecific reactions. Subsequently, the sections were incubated with primary antibodies (Table 2) overnight at 4°C. After being washed in Tris-buffered saline, the sections were incubated with horseradish peroxidase-labeled polymer (Dako, Tokyo, Japan). For the detection of laforin, biotinylated rabbit anti-goat IgG antibody (KPL, Gaithersburg, MD) was used for the secondary antibody, followed by incubation with peroxidase-conjugated streptavidin (Dako). The reaction products were visualized with 0.05% 3-3’-diaminobenzidine and 0.03% hydrogen peroxide in Tris-HCl buffer, and then the sections were counterstained with hematoxylin. Antibody diluent and an irrelevant antibody were used as negative controls. 22
Antibodies Used for Immunohistochemistry.
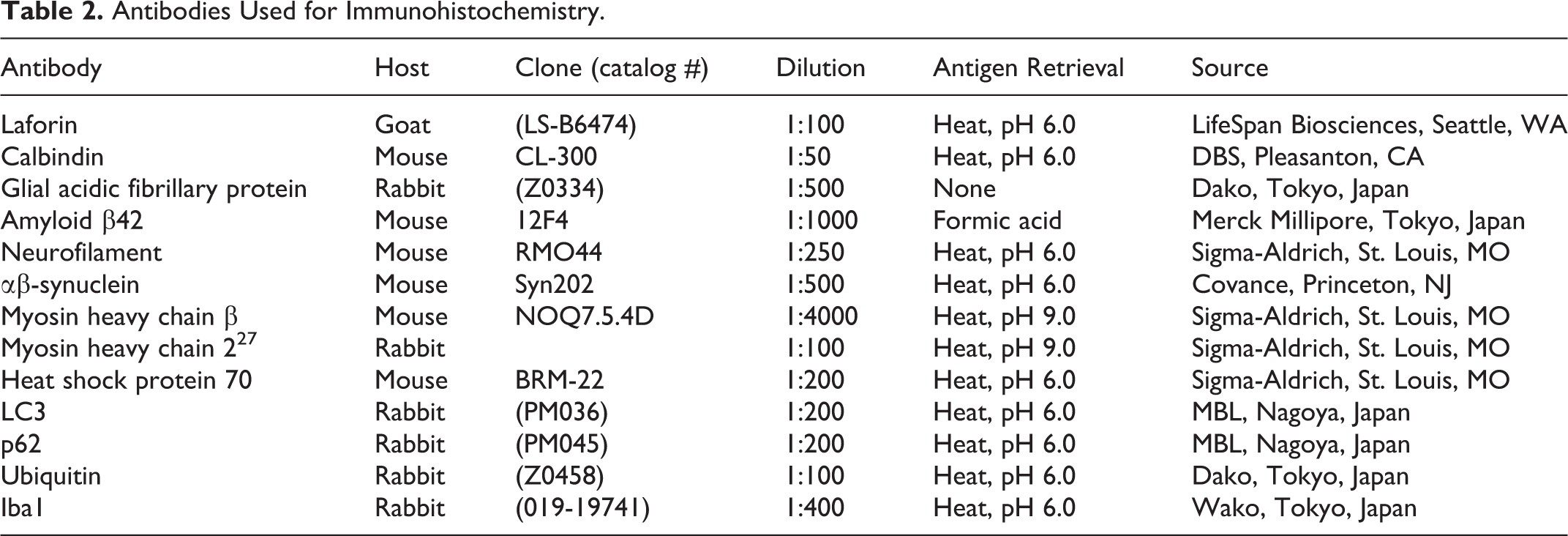
Double-Labeling Immunofluorescence
Deparaffinized sections underwent the same immunohistochemical procedure as described above. After incubation with 2 of the primary antibodies overnight at 4°C, the sections were washed with TBS, incubated with a corresponding secondary antibody for 1 h at 37°C, and then mounted with Vectashield (H-1500, Vector Laboratories, Burlingame, CA). The primary antibodies used were as follows: goat anti-laforin antibody, rabbit anti-GFAP antibody, mouse anti-neurofilament antibody, and rabbit anti-ubiquitin antibody. The secondary antibodies used were as follows: ALEXA 488-conjugated donkey anti-goat IgG (1:100, Invitrogen, Eugene, OR), ALEXA 594-conjugated donkey anti-mouse IgG (1:100, Life Technologies, Eugene, OR), and ALEXA 594-conjugated donkey anti-rabbit IgG (1:100, Life Technologies).
Results
Clinical Manifestation and General Pathological Findings
The initial clinical sign in all 4 dogs was myoclonic seizure appearing between ages 7 and 9 years (Table 1). Magnetic resonance imaging revealed no significant findings. At neurological examination, myoclonic seizure was induced by the menace test or photostimulation. As the disease progressed, the dogs exhibited visual disturbance. However, myoclonic episodes were controlled with antiepileptic drugs (Table 1). PCR analysis for the EPM2B gene revealed that all dogs were homozygous for the expanded EPM2B gene (Fig. S1). The dogs died between 14 and 16 years of age, however the clinical signs did not suggest neurologic disease as a cause of death. Disease duration was 8 to 9 years. The lateral ventricles were moderately enlarged and mild atrophy of the cerebral and cerebellar gray matter was observed on the cut surface of the fixed brains. Upon general histopathological examination, severe suppurative bronchopneumonia was the major finding in 3 cases (cases 1, 2, and 3), and lymphocytic plasmacytic interstitial nephritis and cystitis were the major finding in 1 case (case 4). No other significant histopathological findings other than polyglucosan body formation, described later, were observed in the 4 dogs.
Morphology and Distribution of Polyglucosan Bodies
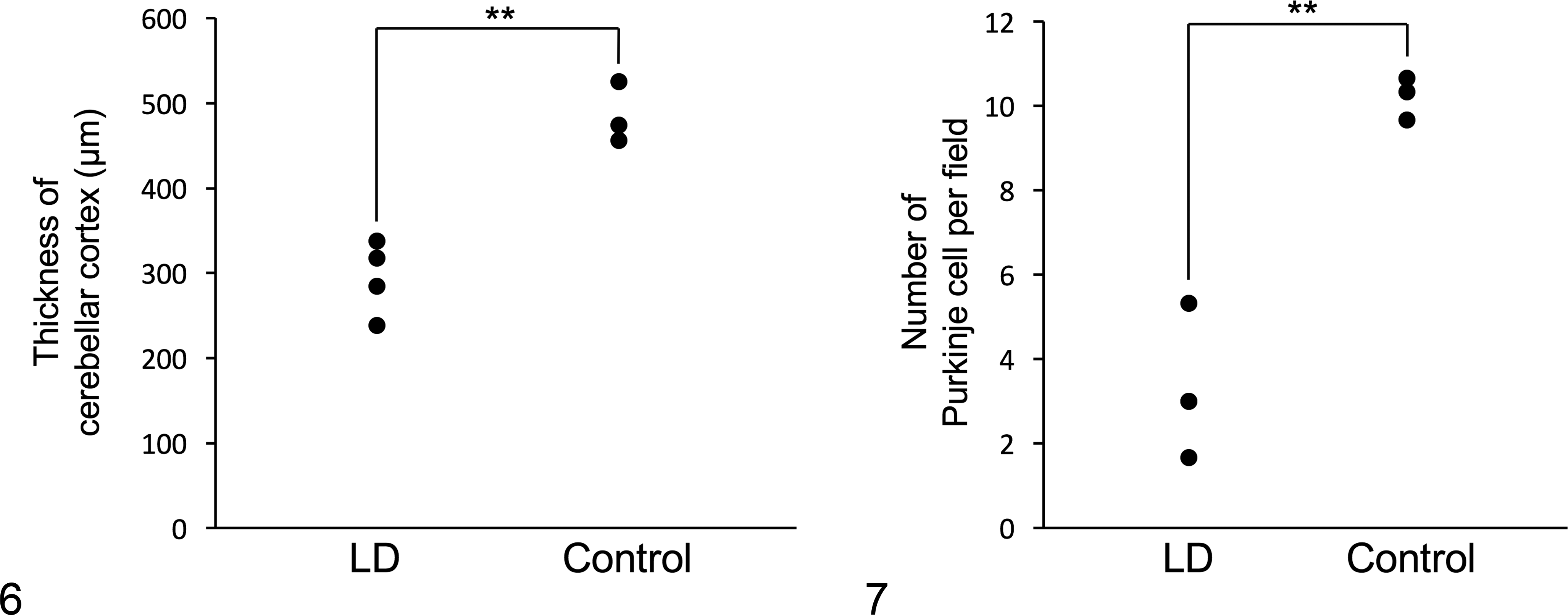
On the hematoxylin and eosin-stained sections, basophilic round polyglucosan bodies with a diameter of 5–20 μm were frequently observed in the gray matter of the central nervous tissues, most prominently in the atrophic cerebellar cortex (Fig. 1). The polyglucosan bodies had dense central cores and faintly stained outer layers, occasionally showed a radial appearance, and were positive for PAS and Alcian blue (Figs. 2 and 3). Immunohistochemistry for calbindin revealed that the polyglucosan bodies were located in the cell bodies and dendritic processes of the Purkinje cells (Fig. 4). Some Purkinje cells were lost, and in such areas, few polyglucosan bodies were engulfed by Iba1-positive phagocytic cells. The thickness of cerebellar cortex and the number of Purkinje cells per field were significantly reduced in LD compared to non-LD controls (Figs. 4–7).
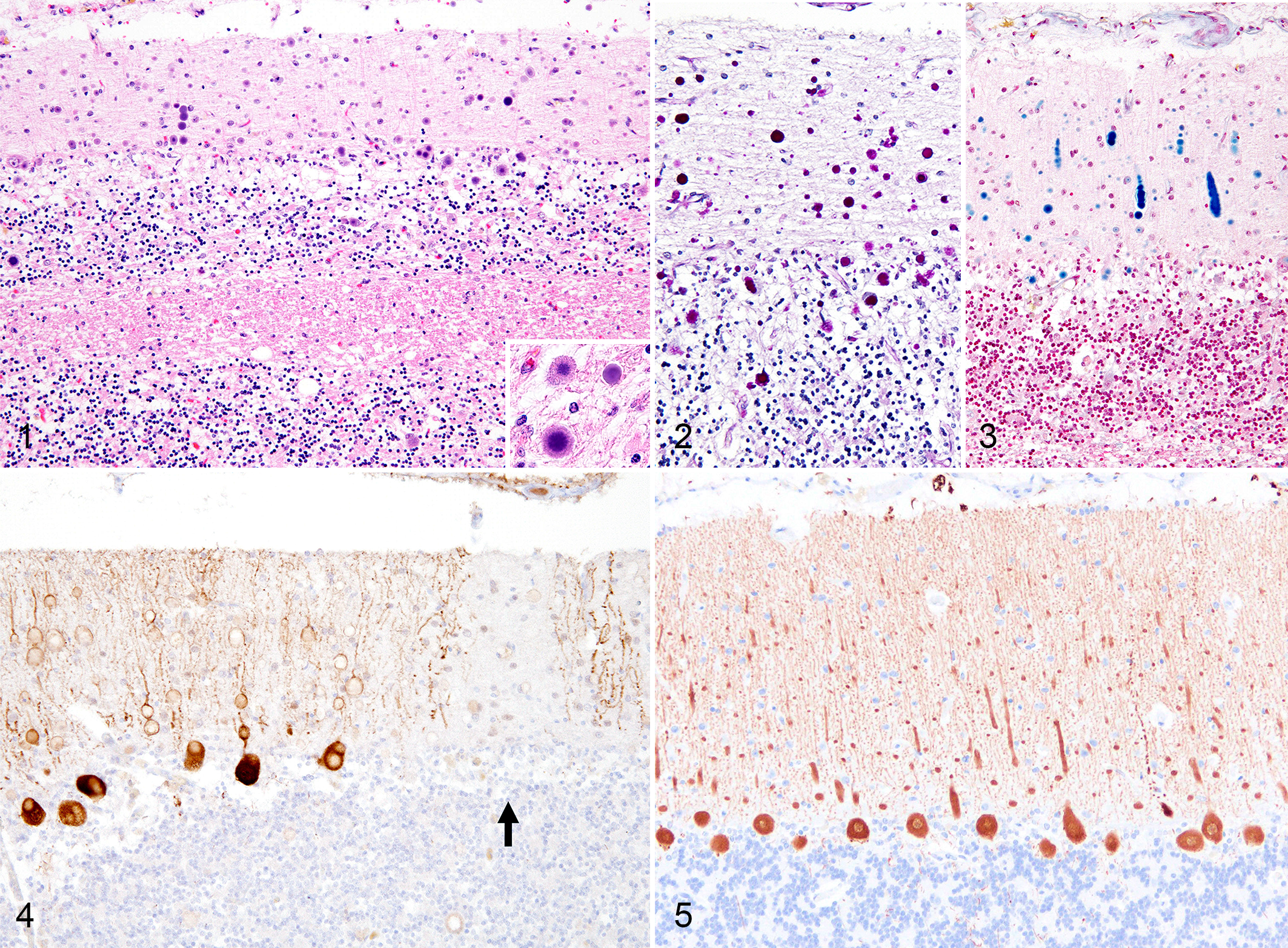
In neuronal tissues, PAS-positive polyglucosan bodies were observed in the cerebral cortex, basal ganglia, diencephalon, pons, cerebellar cortex, choroid plexus, medulla oblongata, the gray matter of the spinal cord, retina, trigeminal nerve, and the sensory ganglia in all cases examined (Figs. 8–10). Secondary changes such as astrocytosis or microgliosis were very limited or absent. In nonneural tissues, PAS-positive polyglucosan bodies were observed in cardiomyocytes in the heart (4/4 cases), myofibers in skeletal muscle (3/3 cases), hepatocytes in the liver (3/4 cases), epithelial cells within apocrine sweat glands (2/4 cases), and smooth muscle cells in the urinary bladder (4/4 cases). However, PAS-positive polyglucosan bodies were very few in number in the apocrine sweat gland compared to other tissues. There were 1 or 2 polyglucosan bodies in 1 slide (approximately 3 cm in length) of skin tissue.
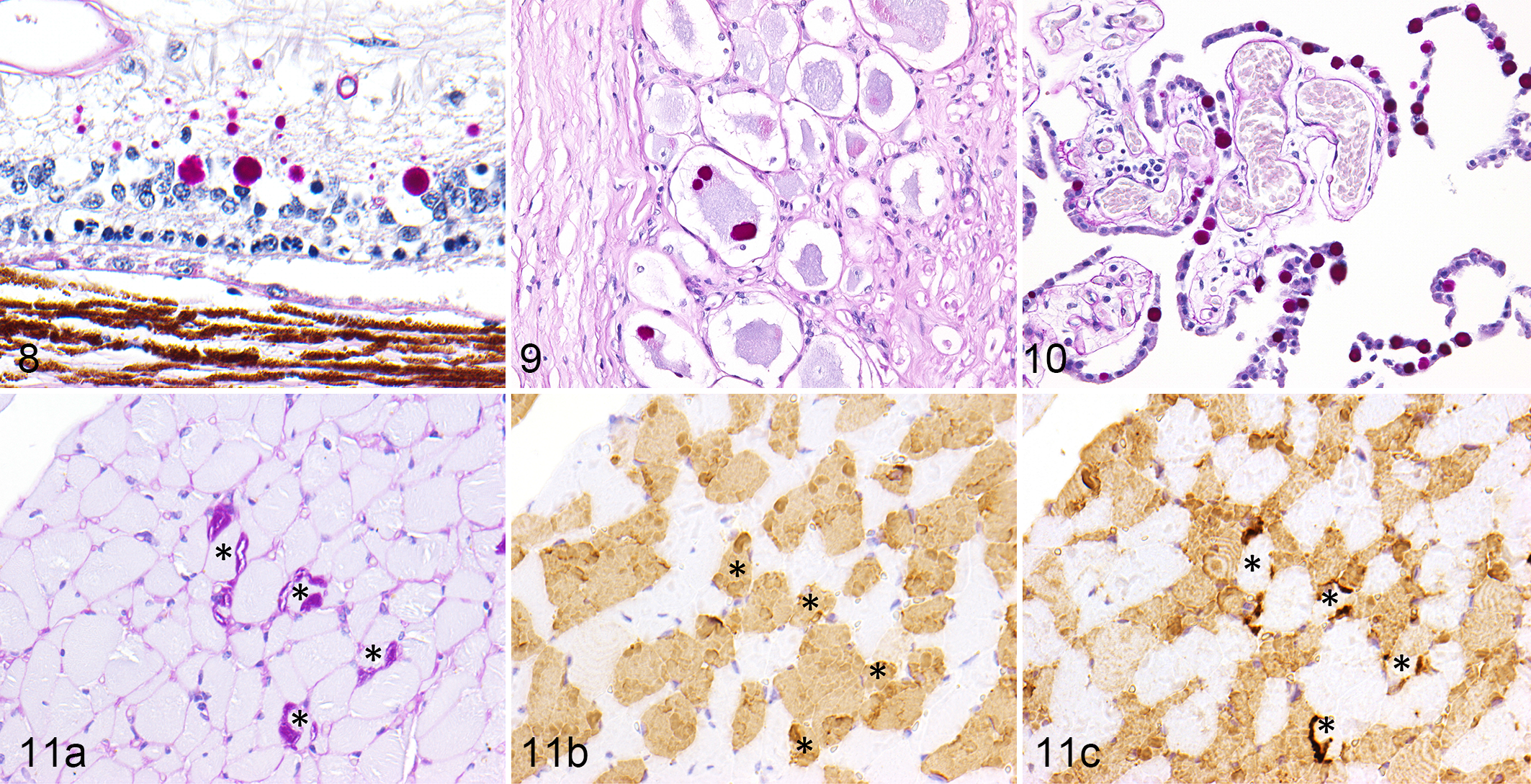
Of the skeletal muscles examined, PAS-positive polyglucosan bodies were observed mainly in the longissimus, but also in the triceps brachii, lateral rectus, diaphragm, and quadriceps femoris. Immunohistochemistry of consecutive sections revealed that PAS-positive polyglucosan bodies were observed only in myosin heavy chain β-positive type 1 fibers and not in myosin heavy chain 2-positive type 2 fibers (Figs. 11a–11c). The polyglucosan bodies were not observed in the esophagus or oblique abdominal muscle.
Immunostaining of Polyglucosan Bodies and Protein Deposits
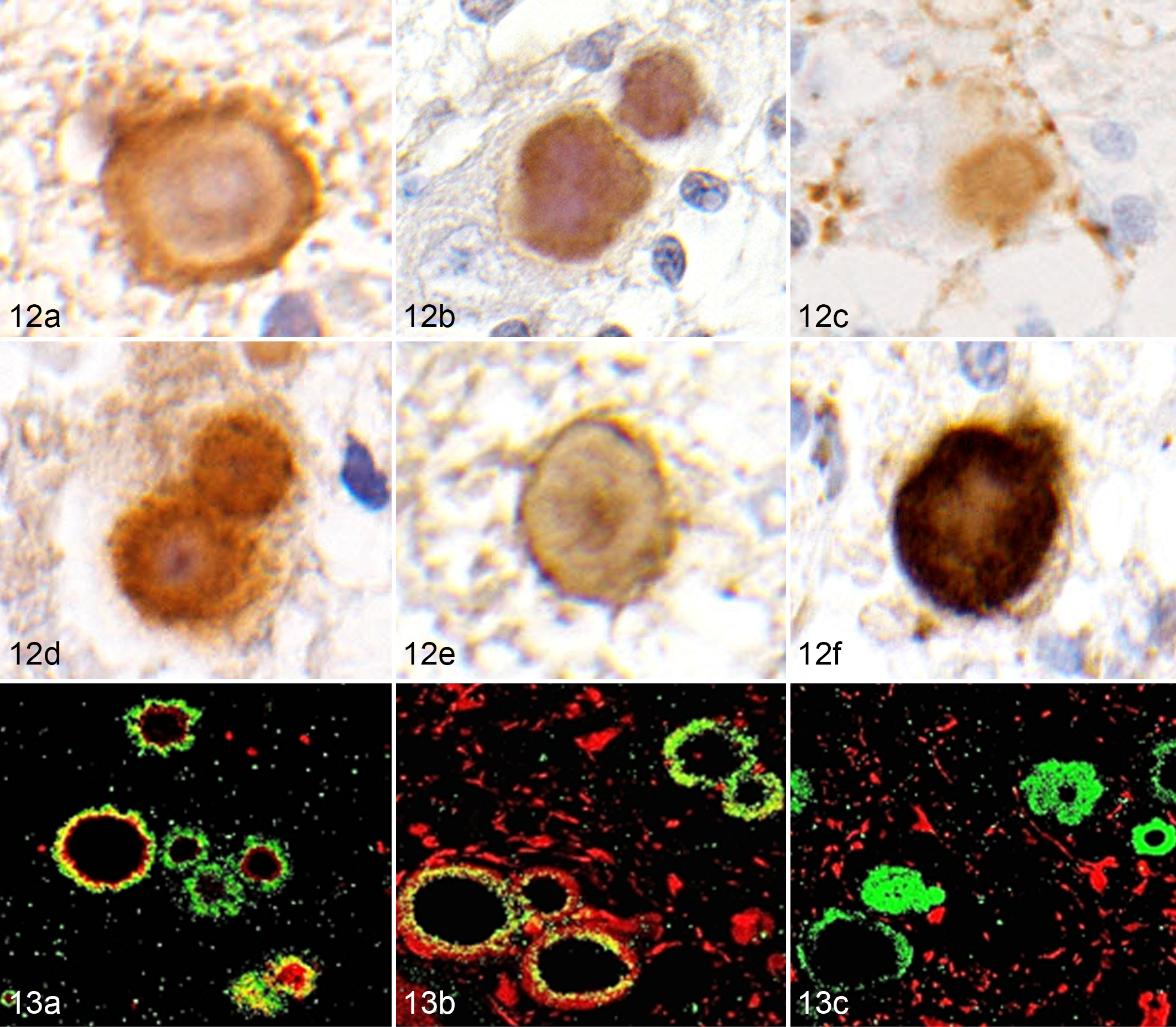
Polyglucosan bodies in the cerebrum and cerebellum were strongly positive for laforin, hsp70, αβ-synuclein, ubiquitin, LC3, and p62 (Figs. 12a–12f). Colocalization of laforin and ubiquitin was confirmed using double immunofluorescence (Fig. 13a). Laforin-positive polyglucosan bodies were observed in neurofilament-positive neurons but not in GFAP-positive astrocytes (Figs. 13b and 13c). Few polyglucosan bodies were observed in the brains of non-LD miniature dachshunds. On serial sections, some of those polyglucosan bodies were positive for laforin, hsp70, αβ-synuclein, ubiquitin, LC3, and p62.
In addition to the polyglucosan bodies, Aβ42 deposits were observed in all cases. Aβ42 was deposited in the cerebral cortex as plaques and in the vascular walls of the meninges and cerebral and cerebellar cortices.
Discussion
Repeat expansion in the EPM2B gene is the only identified cause of LD in the dog. 15 Clinical onset of canine LD with EPM2B repeat expansion occurred at 6–9 years of age. The disease duration for the EPM2B mutation was comparable between canine LD and human LD. 25 In the present study, magnetic resonance imaging of the brain did not show significant change, probably because it was performed at the early stage of disease. 13 Enlargement of lateral ventricles at necropsy (ie, the end stage of the disease) is likely caused by both neurodegeneration from LD and also aging. The significant atrophy of the cerebellar cortex and decrease of Purkinje cells indicate severe neurological impairment in canine LD. However, unlike in humans, canine LD is not a fatal condition because life expectancy is not affected and development of typical incidental age-related neuropathological findings, such as Aβ deposits, accompany the LD in the brain. 36
Diagnosis of LD is confirmed by clinical signs (myoclonic seizures), results of genetic tests (homozygous mutations in EPM2A or EPM2B), and histopathological findings (polyglucosan bodies). However, since there are other diseases that may cause myoclonic seizures, and as there are LD cases that are negative for EPM2A and EPM2B mutations, the detection of polyglucosan bodies (Lafora bodies) is important for the diagnosis of LD. Polyglucosan bodies were detected in the brain and liver biopsy samples of presymptomatic children with LD, 2 and the liver function test has been shown to be an indicator for disease onset. 12 Moreover, histopathological detection of polyglucosan bodies in skin and muscle biopsy samples are the least invasive methods for diagnosing LD in the early stages of the disease. 16,20 In the present canine study, polyglucosan bodies were intensely distributed in the central nervous system, and the neurons contained abundant polyglucosan bodies. In addition, the polyglucosan bodies were also observed in the skeletal muscle, the smooth muscle of the urinary bladder, the liver, and less frequently, in the skin. The skeletal muscle and the skin are the least invasive biopsy site for antemortem histopathological diagnosis of canine LD. However, the numbers of polyglucosan bodies were few in these tissues, and thus genetic analysis would be more reliable and efficient in diagnosing the disease. In the quadriceps femoris muscle of laforin-deficient and malin-deficient mice, polyglucosan bodies develop in type 2 fibers but not in type 1 fibers; this finding may be associated with the difference in glycogen metabolism between the fiber types in mice. 31 However, in the present canine study, the polyglucosan bodies were observed specifically in type 1 fibers and not in type 2 fibers. The triceps brachii, quadriceps femoris, and diaphragm were the most affected, in accordance with the proportion of type 1 fibers in the muscles. Species differences in the development of polyglucosan bodies in the skeletal muscle should be considered.
Although the pathomechanism of LD is still not fully elucidated, dysfunction of laforin and/or malin has been indicated in studies using an LD disease model in mice and in human patients. Malin functions as a ubiquitin ligase and ubiquitinates proteins related to glycogen metabolism, such as the protein targeting to glycogen (PTG) and neuronatin (NRT), and induces proteasome-dependent degradation of those proteins (Fig. 14). 26,34,35 NRT activates glycogen synthase (GS) and regulates glycogen synthesis. Therefore, decrease or dysfunction of malin results in abnormal glycogen metabolism leading to polyglucosan body formation. Malin also ubiquitinates laforin and promotes its degradation. 11 It has been shown that expression of malin is significantly decreased in dogs that are homozygous for the EPM2B repeat expansion. 17 In addition, the structure of the malin protein is significantly altered due to the repeat expansion. As demonstrated in malin-deficient LD model mice, 7 the accumulation of laforin and the formation of polyglucosan bodies in the affected dog may be due to the depletion and/or dysfunction of the malin protein (Fig. 15). However, immunohistochemistry for malin was not performed in the present study because of the unavailability of an antibody against the canine malin protein.
Laforin is highly conserved in mammalian species, binds glycogen via the CBM, and is involved in glycogen dephosphorylation (Fig. 14). 4 Increased laforin and laforin-binding to glycogen within the cell also contribute to the formation of polyglucosan bodies in malin-deficient LD model mice, 30 and possibly in canine LD.
In addition, laforin positively regulates autophagy by increasing the expression of LC3, a protein involved in the formation of autophagosomes (Fig. 14). 1 In the autophagy process, p62 interacts with LC3 and regulates the autophagic response, and then is removed by autophagy. 32,33 In the present canine study, the accumulation of LC3 and p62 was observed in the polyglucosan bodies, indicating that altered autophagy-related protein accumulation may be involved in the pathomechanism of canine LD (Fig. 15).
In both EPM2A- and EPM2B-related human LD, HSP70, a chaperone protein, and ubiquitin are present in the polyglucosan bodies. 23 Laforin and malin, together with HSP70, compose a functional complex and suppress the cellular toxicity of misfolded proteins such as α-synuclein (Fig. 14). 10 The hyperphosphorylation and aggregation of tau protein has been demonstrated in laforin-deficient mice. 21 An in vitro study has shown that proteasomal impairment in a cell results in the accumulation of laforin and malin, together with ubiquitin. 18 The overload or impairment of the ubiquitin proteasome system may result in the aggregation of toxic misfolded proteins and the recruitment of molecular chaperones (Fig. 15).
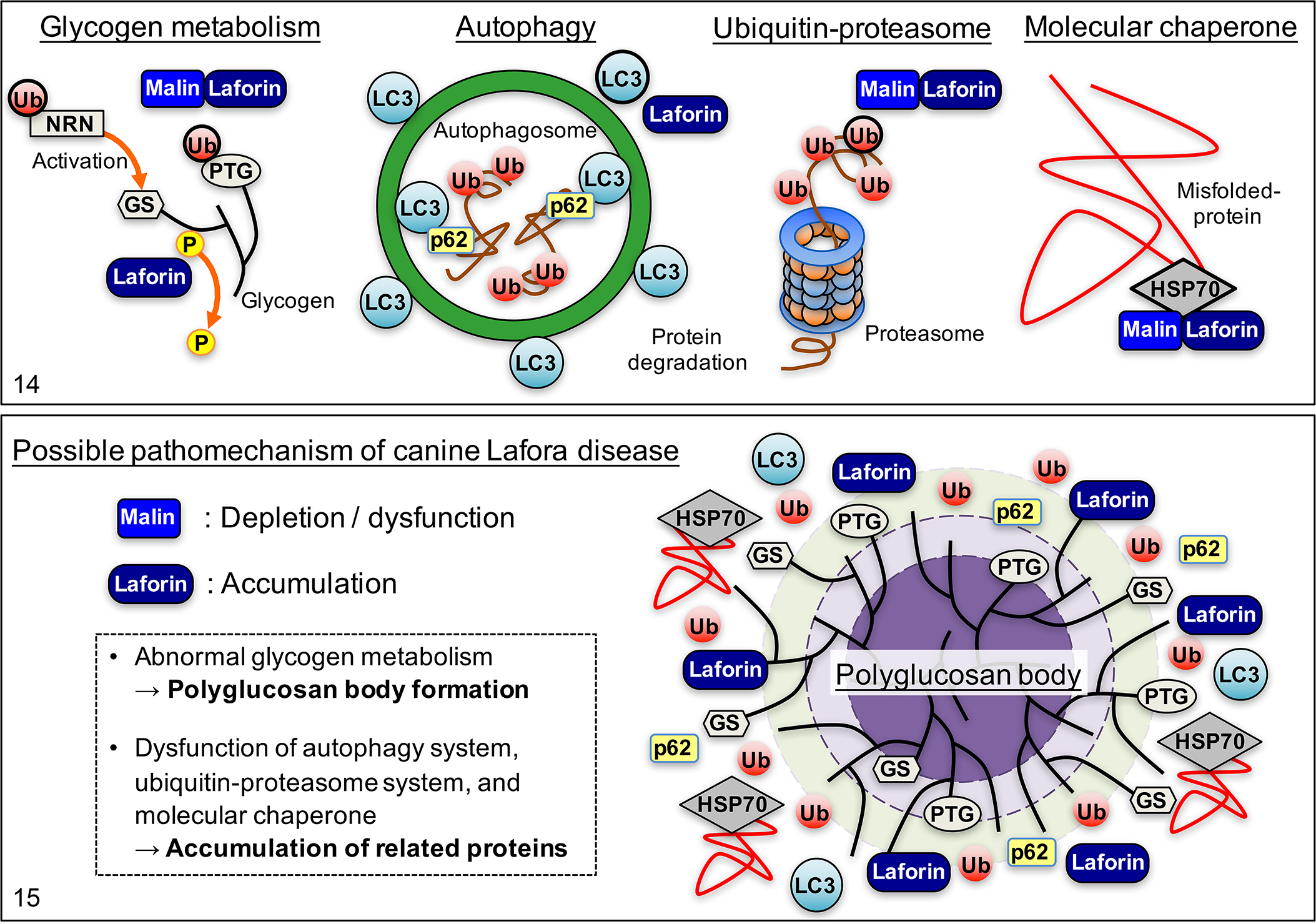
In conclusion, disordered protein degradation (ubiquitin-proteasome and autophagy) and glycogen metabolism are likely to be involved in the pathomechanism of LD in the dog as well as in humans. Since some of the polyglucosan bodies in the brains of non-LD aged dogs were also positive for the immunohistochemical markers that were examined, polyglucosan body development in LD and aging may involve similar mechanisms, although the degree of polyglucosan body formation is significantly different. Although the repeat expansion of the EPM2B gene is specific to the dog, the underlying pathomechanism and morphology of the polyglucosan bodies in canine LD are comparable to that in human LD. Canine LD may be a promising animal model for disease modifying therapy targeting the autophagy and ubiquitin proteasome systems. However, evaluating the therapeutic effect on prolonged survival may be difficult in the canine disease model, because the disease does not affect longevity. Also, there are some variations in the formation of polyglucosan bodies among animal species, such as in the skeletal muscle of mice and dogs. In addition, unlike human LD patients, canine LD cases have the same life expectancy as unaffected dogs and thus also develop age-related changes of the brain.
Supplemental Material
Supplemental Material, DS1_VET_10.1177_0300985818758471 - Accumulation of Laforin and Other Related Proteins in Canine Lafora Disease With EPM2B Repeat Expansion
Supplemental Material, DS1_VET_10.1177_0300985818758471 for Accumulation of Laforin and Other Related Proteins in Canine Lafora Disease With EPM2B Repeat Expansion by James K. Chambers, Atigan Thongtharb, Takanori Shiga, Daigo Azakami, Miyoko Saito, Masumi Sato, Motoji Morozumi, Hiroyuki Nakayama, and Kazuyuki Uchida in Veterinary Pathology
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.