
Abstract
Human idiopathic inflammatory demyelinating diseases (IIDD) are a heterogeneous group of autoimmune inflammatory and demyelinating disorders of the central nervous system (CNS). These include multiple sclerosis (MS), the most common chronic IIDD, but also rarer disorders such as acute disseminated encephalomyelitis (ADEM) and neuromyelitis optica (NMO). Great efforts have been made to understand the pathophysiology of MS, leading to the development of a few effective treatments. Nonetheless, IIDD still require a better understanding of the causes and underlying mechanisms to implement more effective therapies and diagnostic methods. Experimental autoimmune encephalomyelitis (EAE) is a commonly used animal model to study the pathophysiology of IIDD. EAE is principally induced through immunization with myelin antigens combined with immune-activating adjuvants. Nonhuman primates (NHP), the phylogenetically closest relatives of humans, challenged by similar microorganisms as other primates may recapitulate comparable immune responses to that of humans. In this review, the authors describe EAE models in 3 NHP species: rhesus macaques (Macaca mulatta), cynomolgus macaques (Macaca fascicularis), and common marmosets (Callithrix jacchus), evaluating their respective contribution to the understanding of human IIDD. EAE in NHP is a heterogeneous disease, including acute monophasic and chronic polyphasic forms. This diversity makes it a versatile model to use in translational research. This clinical variability also creates an opportunity to explore multiple facets of immune-mediated mechanisms of neuro-inflammation and demyelination as well as intrinsic protective mechanisms. Here, the authors review current insights into the pathogenesis and immunopathological mechanisms implicated in the development of EAE in NHP.
Keywords
Human idiopathic inflammatory demyelinating diseases (IIDD) of the central nervous system (CNS) comprise a broad spectrum of disorders with multiple differences in their clinical appearance. IIDD have been traditionally described as acquired idiopathic disorders characterized by myelin loss that occurs on a background of inflammation with relative preservation of axons. 87,91 Their respective etiologies remain mysterious, but epidemiologic and genomic studies point to immune dysfunctions. 11,21,48 Among IIDD, multiple sclerosis (MS) is the most common and extensively studied human demyelinating disorder for which considerable efforts have been made to understand its pathophysiology and ultimately find a cure. 78 With this goal in mind, experimental autoimmune encephalomyelitis (EAE) models are commonly exploited to study IIDD. While EAE models could involve virtually any mammal, it is mouse EAE models that have defined the bulk of our present knowledge concerning immunopathogenic mechanisms of IIDD, with EAE in nonhuman primates (NHP) representing only a small fraction (<10%) of these studies. EAE in rodents has been used to develop therapeutic molecules presently used to treat relapsing-remitting MS (RRMS), proving the pertinence of this model. 7,22 However, some of these immunotherapies valuable in mouse models have proven ineffective or even detrimental when tested in MS patients. 103,121 Differences in immune responses between specific pathogen-free (SPF) mice and humans may represent a major obstacle to the translation of immune therapies. The immune system of NHP, challenged by microorganisms similar to those of humans from childhood to adulthood, may recapitulate comparable immune responses to that of humans. Therefore, primate models of EAE may help to further refine our understanding of the pathophysiology of IIDD and close the gap between mouse EAE models and human patients. Moreover, the genetic similarity between NHP and humans, with great homology of protein sequences and functions, makes NHP more appropriate to test the properties of therapeutic molecules.
In this review, we describe EAE models in 3 NHP species, rhesus macaques (Macaca mulatta), cynomolgus macaques (Macaca fascicularis), and common marmosets (Callithrix jacchus), evaluating their contribution to the understanding of human IIDD. The spectrum of EAE models in NHP discussed in this review may be helpful to: (1) elucidate underlying mechanisms of IIDD, (2) identify markers of disease onset and progression, (3) establish imaging protocols to follow inflammatory and/or demyelinating lesions, and (4) test preclinical therapeutic protocols.
Human Idiopathic Inflammatory Demyelinating Diseases
IIDD represent a broad spectrum of CNS disorders that can be differentiated on the basis of patient history, clinical course, neurological examination, neuroimaging of the brain, as well as laboratory and pathologic features. 98 Although rarely required, biopsies are performed to exclude other diseases (most often tumors) and establish the final diagnosis of IIDD. The spectrum includes monophasic, multiphasic, and progressive disorders, ranging from highly localized forms to multifocal or diffuse variants. 41 This group of CNS IIDD includes MS and its acute variant the Marburg form of MS, neuromyelitis optica (NMO), Balo’s concentric sclerosis, acute disseminated encephalomyelitis (ADEM), and its hyperacute variant acute hemorrhagic leukoencephalitis (AHL). 41
MS is a chronic disease, and it is the most common cause of disability of young adults, which affects up to 2 million people worldwide. 21 In terms of clinical course, there are several MS subtypes. The most frequent one is RRMS, which has episodes of neurological disability separated by periods of remission. Another is secondary progressive MS (SPMS), which represents a later step of RRMS with unremitting gradual neurological deterioration (disease progression). Also, primary progressive MS (PPMS) is a rare form that manifests as unremitting gradual neurological deterioration that lacks intermediary remissions. An even rarer variant, called progressive-relapsing MS, is seen when a few acute relapses are superimposed on the gradual primary progressive course.
Possible Triggers of Idiopathic Inflammatory Demyelinating Diseases
The heterogeneity of human IIDD may suggest different causes, but the etiology and pathophysiology of these disorders remain poorly understood despite extensive investigations. 19,27,44 The general consensus is that an autoimmune reaction occurs against myelin sheaths and oligodendrocytes, contributing to the central part of the pathogenesis. 98 However, it is still not proven whether myelin instability (ie, myelin sheet decompression, viewed by electronic microscopy as separation of myelin lamellae) in MS is a direct consequence of autoimmune inflammation, as observed in EAE models, or a secondary consequence of axon degeneration, as observed in Theiler’s murine encephalomyelitis. 88,112
The debate about the nature and sequence of immunopathologic events triggering and exacerbating human IIDD are still ongoing and remain largely speculative, especially regarding the respective roles of genes and environment on the origin and progression of the disease. 3,26 Human genetic studies, epidemiology, and animal experimentation are all important approaches to help clarify IIDD pathogenesis. For instance, studies of monozygotic twins indicate that about 30% of the etiology of MS is genetic. 29 Genome-wide association studies clearly show that there are MS susceptibility regions coding immunoregulatory factors, with a central role given to antigen presentation, NF-κB signaling, and T-cell proliferation. 8,46,48 On the other hand, viral pathogens are known to be the most critical infectious factors triggering IIDD. The evidence mostly supports a role of Herpes viruses, such as human betaherpesvirus 6 (HHV-6), human gammaherpesvirus 4 (also known as Epstein-Barr virus [EBV]), and to a lesser extent human betaherpesvirus 5 (also known as cytomegalovirus [CMV]). 30,55,70 In addition, there are many bacteria thought to provoke autoimmune demyelination, including Mycoplasma pneumoniae, Chlamydophila pneumoniae, Borrelia burgdorferi, and Haemophilus influenzae. 79,120 Furthermore, it was recently suggested that human IIDD, and especially MS, could result from aspects of the human lifestyle in modern and developed countries, such as immune imprinting by diet from early stages in life, an immune exacerbation by high fat and/or salt intakes, as well as an omnivorous alimentation with a prominent carnivorous diet. 52,102,118
Development of EAE Models in Rodents and NHP
The idea that the development of certain neurological diseases may have an autoimmune pathogenesis was first proposed in 1927 by Eduard Glanzmann as an explanation for the development of encephalomyelitis after certain viral infections such as chickenpox and smallpox as well as after rabies vaccination. 36 Indeed, at the end of the 19th century, Pasteur’s rabies vaccines were prepared as emulsions of spinal cords from rabbits infected with an attenuated rabies virus, and injection of this preparation led to neurological complications in 40 out of 107 of his patients. This was at that time attributed to incomplete virus inactivation but later found to be ADEM. 110 In fact, in 1933, Thomas Rivers showed that administration of noninfected foreign brain extracts into a rabbit’s brain could induce paralysis, which was then replicated in rhesus macaques. 113 Neurological deficits found in the NHPs used in these experiments were correlated with histological lesions of encephalomyelitis, including perivascular leucocyte infiltration, necrotic and hemorrhagic areas, and demyelination. 93 This established the first EAE models, their association with an immune dysfunction, and myelin as the disease trigger. 10,53
Active EAE is commonly elicited through immunization with myelin antigens, which leads to combined activation of innate and adaptive immunity. For decades, EAE models were induced with whole brain or white matter homogenate with a strong adjuvant, such as complete Freund’s adjuvant (CFA), to accelerate and potentiate disease onset. 53 Since these first experiments, EAE has been induced by a variety of methods, including immunization with purified white matter proteins (or derived peptides) such as myelin basic protein (MBP), proteolipid protein (PLP), myelin oligodendrocyte glycoprotein (MOG), or other myelin proteins. Additionally, proteins from astrocytes, neurons, and cerebral endothelial cells have been also used. 32,96,111,114 More recently, it was shown that EAE could be efficiently induced in NHP with MBP or MOG in incomplete Freund’s adjuvant (IFA), an innocuous adjuvant containing mineral oil but lacking bacterial extract. 38
For many years, rodent models of EAE dominated research in autoimmune inflammation of the CNS, and most studies are currently done using C57BL/6 or SJL/J strains. 77,106 For instance, C57BL/6 immunization with the peptide MOG(35–55) in CFA can induce a monophasic form of EAE, characterized by confluent areas of mononuclear inflammatory infiltration and demyelination in the peripheral white matter of the spinal cord. 23 SJL/J mice immunized with the peptide PLP(139–-151) develop a relapsing-remitting disease with perivascular and meningeal lymphocyte and neutrophil infiltration, followed by resolution of the inflammatory infiltrate and at the same time progression of white matter damage, including demyelination and gliosis. 22
Passive EAE is established by transfer of myelin-specific autoreactive T-cells from animals with an actively induced disease to a suitable naive recipient, usually an individual from the same strain. The variety of passive EAE models in mice are typically based on transfer of TH1 cells reacting to myelin epitopes, such as PLP(139–151), MBP(84–102), or MOG(35–55), and these mainly induce inflammation. 63,123 Active and passive EAE models using genetically modified C57BL/6 mice overexpressing proinflammatory mediators (ie, cytokines) are used to study the mechanisms of CNS inflammation. 83 Furthermore, transgenic C57BL/6 mice overexpressing T-cell receptors specific to MOG(35–55) and double-transgenic mice, which in addition to the TCRMOG(35–55) overexpress the heavy chain of anti-MOG IgG, allow the investigation of T-cell and B-cell contribution in the pathogenesis of EAE. 13,60 More recently, transgenic mice expressing MS-associated major histocompatibility complex (MHC) class II haplotypes and human-derived myelin (MBP)-specific TCR have been shown to spontaneously develop EAE. 31
Without doubt, rodent EAE models are vital for studying general concepts as well as specific mechanisms of autoimmunity; however, these models suffer from numerous limitations in regard of pathophysiology of IIDD. The most important component in the adjuvant CFA is heat-inactivated Mycobacterium tuberculosis, which always induces a prominent CD4+ TH1 response by activating certain Toll-like receptors. 100 This leaves little room for variability in disease pathways and certainly does not reflect the heterogeneous nature of MS. The roles of CD8+ T-cells, which predominate in MS lesions and show clonal expansion, have been difficult to study using conventional rodent EAE models. 6,47 Furthermore, EAE in rodents is mainly a disease of subpial spinal cord white matter, whereas MS is mainly a brain disease with prominent demyelination of the cerebral and cerebellar cortex. The presence of cortical lesions in rodent EAE has been difficult to discern and is therefore not well studied. 89 Nonetheless, rodent EAE models have contributed enormously to our understanding of autoimmunity and neuroinflammation and permitted the development of certain medicines for RRMS treatment (eg, glatiramer acetate, natalizumab, or FTY720). 22 For the most part, however, these models have proven to be poor predictors of treatment success in MS, particularly concerning the manipulations of inflammatory cytokines such as IL-12, IL-23, IFN-γ, and TNFα. 65 The majority of studies in rodent EAE models involve a limited number of genetically inbred mouse or rat strains, which are raised under SPF conditions. 106 As such, the immune system of these animals is not exposed to similar environmental factors and pathogens, as is the case for the human immune system. 17
Reducing the gap between EAE and IIDD could be achieved by creating new and refined EAE models in humanized mice or switching to a species more closely related to humans, such as NHP. The translation of scientific or therapeutic principles developed in rodents to NHP models may be crucial to assess in the relevance to human IIDD. Importantly, only NHP models allow the preclinical evaluation of molecules such as humanized antibodies. 101,103 Among NHP species, a particularly interesting aspect of marmosets is the stable bone marrow chimerism between twin siblings caused by the sharing of the placental blood stream. Consequently, the immune system of twins is educated in the same thymic environment, making twins immunologically more similar than siblings from single births. 81 This chimerical bone marrow status is highly advantageous for placebo-controlled studies designed to test potential therapeutics as the fraternal twin makes an ideal control. A benefit of such a paired approach is that it reduces the number of animals needed for research.
EAE in NHP
EAE in NHP is clinically heterogeneous with variable development times, clinical course, and severity, depending on which primate species, immunogen (eg, MOG), and adjuvant (eg, CFA) are used. Immunization methods established in mouse models, which use whole brain homogenate or specific white matter proteins combined with CFA, produce a severe, acute, and monophasic disease. However, recently developed NHP EAE models using recombinant human MOG (rhMOG) or MOG peptides in association with IFA can result in a milder disease, with monophasic and polyphasic clinical forms, and better mimic human IIDD. For example, chronic lesions observed in the common marmoset have similarities with the chronic active lesions of MS. 104,109 Thus, we have chosen to review these models more in detail.
NHP immunized with rhMOG/IFA often show a heterogeneous clinical course and disease severity. Chronic polyphasic EAE with multiple relapses and improvements is the main clinical hallmark of the common marmoset EAE model, whereas the rhesus macaque invariably develops a severe acute disease in less than half of immunized animals. Cynomolgus macaques develop EAE with variable clinical presentations, including monophasic fulminant and polyphasic mild forms. 38 Although the biological reasons behind the variability in EAE outcomes in the cynomolgus macaque remain unknown, several factors, including concomitant viral infections (eg, upper respiratory tract infections or persistent herpes virus infections) or individual antigen repertoire (especially of MHC-I genes) are suspected to influence the onset and clinical presentation of EAE. 12,38
IIDD in humans can comprise a wide range of clinical symptoms, including weakness, paresthesia or focal sensory loss, optic neuritis, diplopia, ataxia, and vertigo. Other manifestations can include painful muscle spasms, trigeminal neuralgia, fatigue and depression, subtle cognitive difficulties, psychiatric disturbances, and seizures. 69 EAE onset in NHP often starts with weakness, hemiparesis, paresthesia, and ataxia, which can rapidly progress to paresis, paralysis, and coma. 38 However, other symptoms such as pain, depression, and cognitive dysfunctions are difficult to evaluate in NHP because of the short duration of the disease and a lack of appropriate evaluation methods for assessment of subtle sensory and cognitive difficulties, depression, and pain.
Lesion onset and development in humans and NHP can be monitored using magnetic resonance imaging (MRI). A typical diagnostic hallmark is the presence of hyperintense lesions on T2-weighted images in periventricular, cortical, and spinal white matter. Acute lesions also show signal enhancement on T1-weighted images after administration of gadolinium, an intravenous contrast agent, and this is due to the acute inflammation accompanying the lesions, which compromises the integrity of the blood-brain-barrier (BBB) and permits gadolinium to cross. 22 Furthermore, MRI allows for longitudinal follow-up of the same lesion to monitor whether there is progression (ie, increase of the lesion volume, gadolinium positivity) or regression (ie, decrease of the volume and reestablishment of BBB integrity), which is associated with partial remyelination, as seen in the common marmoset EAE model and in MS. 61,103,104
CNS Lesion Profile of EAE in NHP
Human IIDD and NHP EAE present with similar macroscopic and histopathologic lesions in the CNS. In humans with MS and ADEM and NHP with EAE, the brain white matter, including cortical white matter, corpus callosum, and subpial white matter, are the most frequent target tissues, and lesions in the white matter of the brain stem and in the spinal cord as well as the optic nerve are less frequent. 38,87 Multifocal areas of necrosis and edema of the white matter are the main macroscopic hallmarks of the majority of NHP EAE lesions and in severe cases of ADEM and MS. 9,87 In contrast to MS but similar to certain monophasic IIDD (ADEM, AHL, NMO), hemorrhagic lesions are often present in NHP EAE, especially in large acute lesions 9 (Figs. 1–4).
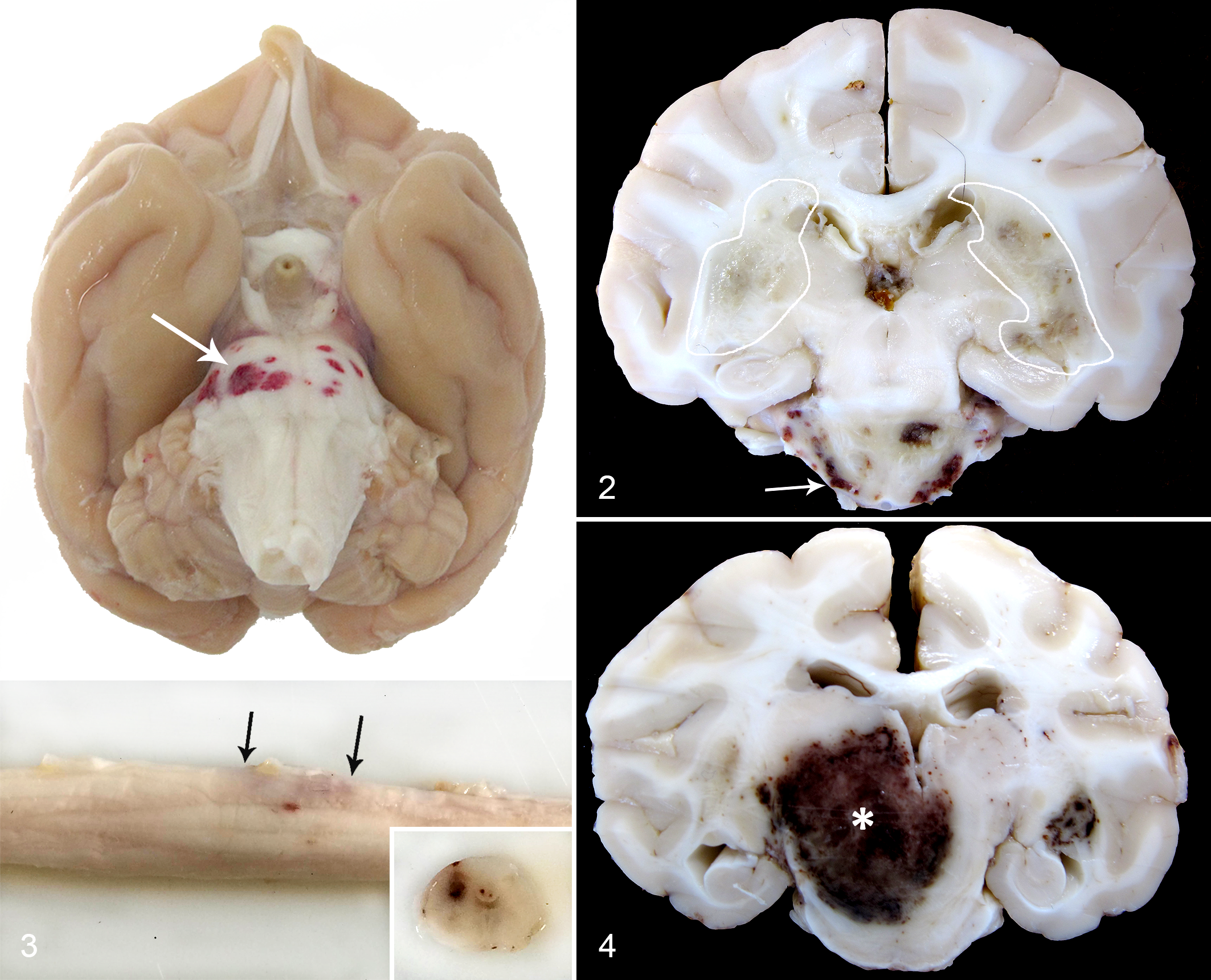
Types of Brain Lesions in NHP EAE
Histologically, EAE in NHP immunized with rhMOG/IFA seems to be a heterogeneous disease with acute lesions described in all 3 NHP species, although they are much more common in cynomolgus and rhesus macaques. These acute lesions are detectable by MRI; they arise shortly after the initial inflammatory and demyelinating event and are almost always correlated with clinical signs of neurological deficits (Table 1). 38,39,71
Comparison of 3 Nonhuman Primate Models of Experimental Autoimmune Encephalomyelitis Induced by Immunization with Recombinant Human Myelin Oligodendrocyte Glycoprotein in Incomplete Freund’s Adjuvant.

Abbreviations: EAE, experimental autoimmune encephalomyelitis; MHC, major histocompatibility complex.
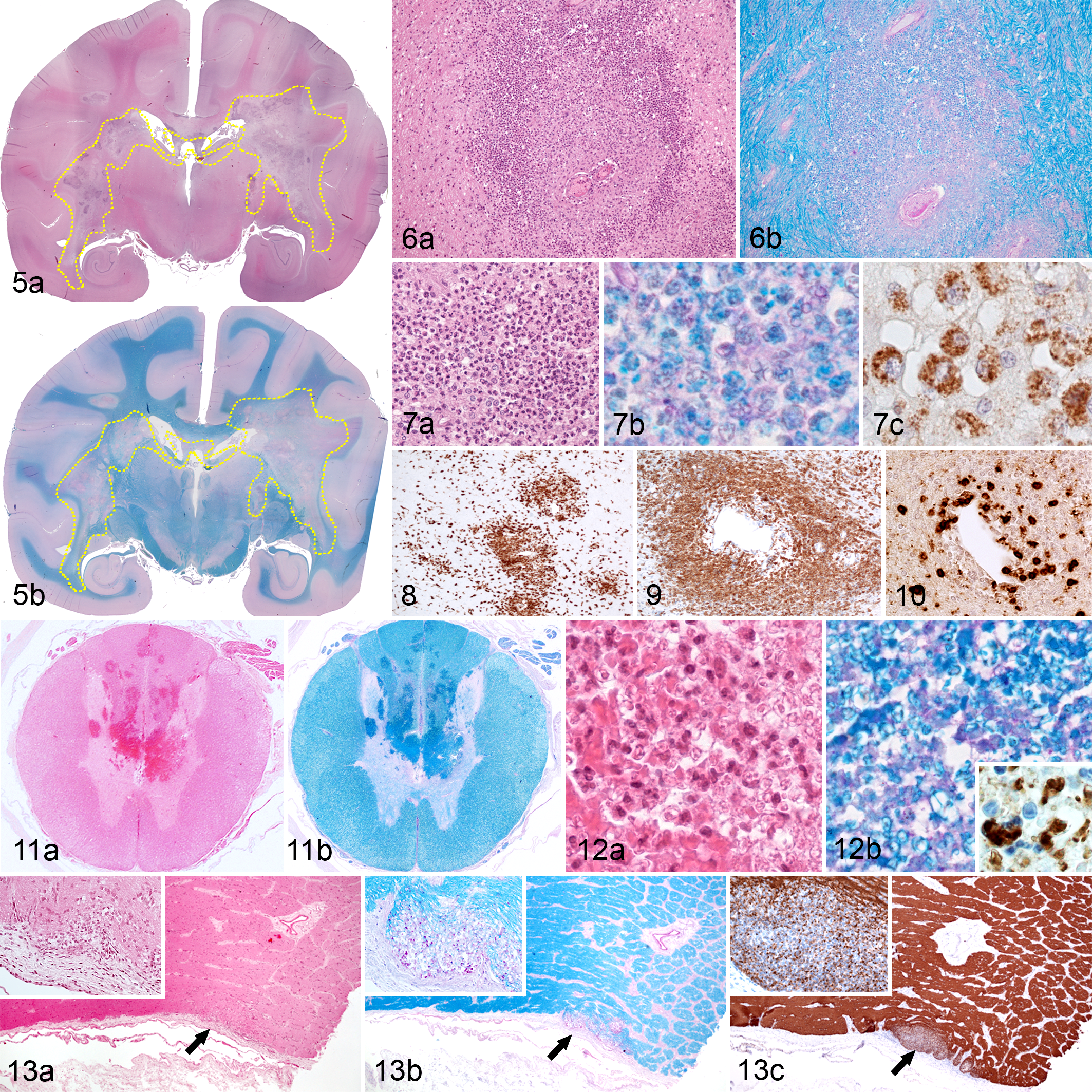
Acute lesions display moderate variability in their size and number. This includes smaller lesions with areas of patchy inflammation and demyelination and larger lesions characterized by 2 distinct compartments: the central necrotic zone and a peripheral demyelinating margin. 9,38 The main histopathological hallmark of the acute lesions is the perivenular distribution of patchy demyelination that starts at the white-grey matter junction. This demyelination is accompanied by an inflammatory infiltrate with high numbers of myelin-laden neutrophils and macrophages, severe loss of oligodendrocytes, and profound perivascular infiltrates composed mainly of macrophages, neutrophils, and fewer T- and B-cells (Figs. 5–10). 38 Similar lesions can be observed in MS patients within acute active plaques, which shows perivenular demyelinating lesions with massive macrophage infiltration. 87 Dissemination of lesions in space and time is the major criterion for MS lesions. For instance, acute NHP EAE lesions, especially in macaques, are disseminated throughout the white matter of the brain; however, all lesions usually seem to be of a similar pathological age, unlike plaques in MS. Furthermore, in contrast to acute lesions of classic-type MS, lesions in NHP EAE are more severe and often contain necrotic and hemorrhagic areas. Severe demyelination and necrosis is one of the key histological features of acute monophasic IIDD (ie, ADEM, NMO, and AHL). The severity of observed lesions in these diseases is comparable to acute lesions in NHP EAE. Furthermore, high number of neutrophils and microhemorrhages is one of the key characteristics of AHL and is frequently seen in humans with NMO. 41,87 Although demyelination is a central event in IIDD and NHP EAE, this lesion is systematically accompanied by a prominent reactive astrocytosis. Uninucleated or multinucleated gemistocytes, mitotic astrocytes, and Creutzfeld-Peters cells (astrocytes with fragmented nuclear inclusions) are often described in acute active lesions of MS and in ADEM. 87 Creutzfeld-Peters cells are not described in acute lesions in NHP EAE; however, astrocytosis and increased glial fibrillary acidic protein (GFAP) expression are always seen in late acute lesions.
Chronic lesions, commonly seen in common marmosets immunized with MOG(34–56)/IFA and more rarely in cynomolgus macaques immunized with rhMOG/IFA, are overall smaller in size with less severe inflammation. 38 In common marmosets sacrificed during the chronic, relapsing-remitting phase of EAE, there are large, sharply defined areas of demyelination, moderate mononuclear cell infiltration, and extensive astrogliosis akin to chronic active plaques of MS. 34 A further important event in chronic lesions of NHP EAE is the partial remyelination. This process starts on the edges of the lesion and is marked by increased 2’3’-cyclic-nucleotide 3’-phosphodiesterase (CNP) expression in clusters of remyelinated axons. 38 Remyelination is well documented in humans with chronic MS lesions, where chronic inactive lesions progressively remyelinate and form so-called shadow plaques after remyelination is complete. 87
Axonal Damage
Axonal injury is often observed in acute lesions but is poorly characterized in NHP EAE. Especially in necrotic lesions, parts of axons are often ruptured or absent, whereas in less severe lesions, these structures are often preserved. Furthermore, in the periphery of larger lesions and those with less inflammation, one can see axonal spheroids and intra-axonal accumulation of proteins, indicating impairment of fast axonal transport. 38 Although axons and neurons are mostly preserved in early MS, the ongoing disease process results in gradual neuroaxonal loss that correlates with patient disability and brain atrophy. 26 Axonal degeneration can be prominent in more acute and destructive diseases such as ADEM, AHL, and NMO. 87 In secondary progressive MS, inflammatory lesions are no longer characteristic; however, a progressive neurological decline is still reported in these patients, which is accompanied by CNS atrophy and increased axonal and neuronal loss. 22 During acute inflammatory demyelination in NHP EAE and in acute lesions of IIDD, axons are probably damaged by the release of toxic inflammatory and nonspecific immune mediators in the lesion (ie, proteases, cytokines, excitotoxins, free radicals). 59 Chronic inflammation in MS results in the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) that probably promote mitochondrial injury, which in turn promotes metabolic stress and energy deficiency, which is critical for neuroaxonal transport and normal function. From the initial site of axonal injury, these degenerative mechanisms can spread backward toward the neuronal cell body (termed retrograde degeneration or neuronal dying back) or toward the distal axon terminal (termed anterograde degeneration or Wallerian degeneration) and can also influence nearby presynaptic and postsynaptic neurons, respectively, eventually leading to neuronal apoptosis or necrosis. 22 Immunopathologic mechanisms of neuroaxonal degeneration are not well characterized in NHP EAE, mainly because of the acute and severe nature of EAE lesions (especially in macaques) and because of the absence of progressive milder inflammatory lesions in these species.
Staging of Brain Lesions in EAE NHP
As mentioned previously, acute lesions in NHP EAE all seem to be of a similar pathological age. Components of destroyed myelin are phagocytized by activated macrophages and neutrophils. After internalization in macrophages, different myelin components have specific rates of degradation; thus, based on their respective presence or absence within macrophages, one can obtain a measure of the demyelinating activity within a lesion. 18 Degradation of minor myelin proteins (ie, CNP, MOG, myelin-associated glycoprotein [MAG]) occurs rapidly, within 1 to 3 days; therefore, immunohistochemical detection of these proteins within macrophages reflects an early stage of demyelination. 87 As in humans with early active plaques, these proteins are frequently observed in early acute lesions in cynomolgus macaques, whereas larger lesions tend to lose MOG reactivity, especially in its center; these lesions can be categorized as late acute lesions. 38 The degradation of major myelin proteins (ie, PLP and MBP) is slower because of their higher quantity, larger size, and hydrophobic nature. 87 PLP, MBP, but not MOG reactivity is observed in late acute lesions of cynomolgus macaques and in chronic lesions of common marmosets and comparable with late active lesions in MS patients. 38,87 Inactive lesions characterized by an absence of PLP reactivity and by the presence of periodic acid-Schiff (PAS)–positive vacuoles in macrophages are rarely seen in chronic lesions of cynomolgus macaques and common marmosets. 38 These PAS-positive vacuoles may correspond to the residue of myelin composed of neutral lipids that are difficult to digest, which in humans may persist up to 6 months. 87
Extracerebral Lesions in NHP EAE
Extracerebral lesions, including lesions of the spinal cord and the optic nerve, are rare in the cynomolgus macaque and the common marmoset. Lesions in the spinal cord are characterized by multifocal, confluent hemorrhages and necrosis of the white and gray matter where injured parts of the tissue are infiltrated by a moderate number of macrophages and neutrophils that phagocytize cellular debris, including myelin (Figs. 11, 12). As in NHP EAE, lesions in the spinal cord are observed with low frequency in MS patients, and similar to the appearance of the cerebral lesions, spinal lesions are characterized by patchy perivenular demyelination infiltrated by numerous macrophages and lymphocytes. 87 In contrast to MS, NMO is a human disorder known to produce demyelinating lesions of the spinal cord with increased severity. As in NHP EAE models, spinal lesions in NMO concern both the grey and the white matter, are often necrotic, and are further characterized by a vasculocentric pattern of immunoglobulin deposition and complement activation. 87
Optic neuritis is another rare consequence of EAE in NHP, characterized by discrete focal demyelinating lesions with few macrophages and lymphocytes (Fig. 13). These lesions are very similar to those observed in MS and NMO patients. 87
Disease Mechanisms of EAE in NHP
Variations of the clinical and pathological appearance of EAE have been observed both between NHP species and between individuals of the same species. These variations, including the incidence, severity, and pathophysiological pathways of EAE in NHP, are determined by 4 major factors: (1) the chosen antigen, including different myelin proteins or peptides recapitulating particular epitopes (eg, MBP, rhMOG, MOG[34–56]); (2) the adjuvants used, as CFA/pertussis toxins, CFA, or IFA; (3) the chosen species and its genetic background that determines the development of innate and adaptive immunity (eg, presence of particular alleles of MHC-II receptors on dendritic cells or MHC-I on somatic cells); and (4) environmental and infectious factors, especially past exposure to viral and bacterial infections (ie, certain herpes viruses, including EBV-like viruses and CMV) 38,95,109 (Table 1).
MOG and Its Implication in the Development of EAE
Proteins comprise 15% to 20% of myelin and include 4 different groups, including immunoglobulins, basic and tetraspan proteins, and enzymes. 40 The integral membrane protein PLP, the extrinsic MBP, and the myelin-associated oligodendrocytic basic protein (MOBP) are the most abundant proteins, while MAG, MOG, and CNP are quantitatively minor constituents. 1,40
White matter antigens, especially myelin glycoproteins, are recognized targets of humoral and cellular immune reactions in CNS demyelination. Among these targets, MOG has been most intensively analyzed, and the results underline its possible role in the pathogenesis of demyelinating diseases and especially of MS. 58,74,92 Indeed, contrary to other myelin proteins such as MBP or PLP, MOG lacks specific central immune tolerance and is particularly prone to induce humoral immunity, which is a central element driving demyelination. 2,25,35,42,76 Thus, this protein is reviewed in more detail.
MOG is a member of the immunoglobulin family and is highly conserved between species. 109 MOG is expressed as a homodimeric complex at the cytoplasmic membrane of oligodendrocytes and the outer lamellae of the myelin sheaths within the CNS. 20 The MOG gene is situated in primates and rodents in the region encoding the MHC-I, and its full length encoded product consists of a 218 amino acids protein (28 kDa). 86,109 While MOG is a minor component with only 0.05% of relative quantity compared to other myelin proteins, its expression is strongly correlated with the process of myelination and therefore represents an important surface marker of oligodendrocyte maturation. 51 Although the function of MOG is poorly understood, there is growing evidence that it possesses several different functions, including cell adhesion, regulation of microtubule stability, and interaction between myelin sheets and immune cells. 51,92,109 It has been shown that myelin interacts with antigen presenting cells (APC) (ie, microglia) in the brain through the interaction between MOG glycans and the receptor DC-SIGN, ensuring anti-inflammatory and regulatory functions. 33 The interaction of MOG with DC-SIGN in the context of simultaneous TLR4 activation results in enhanced IL-10 secretion and decreased T-cell activation and proliferation. On the other hand, exposure of oligodendrocytes to pro-inflammatory cytokines leads to downregulation of glycosylation of MOG, which decreases its interaction with DC-SIGN. It results in the reduction of the anti-inflammatory homeostatic control and leads to subsequent inflammasome activation, T-cell activation, proliferation, and differentiation toward TH17 phenotype. 33
MOG was first identified as a target for demyelinating antibodies in EAE induced by CNS tissue homogenates. 66 MOG appeared to be a crucial factor for the development of EAE in NHP as it was shown that marmosets immunized with myelin from MOG-deficient mice develop only a mild form of disease. 50 Furthermore, several experimental studies have shown that the extracellular domain of MOG is highly encephalitogenic and induces both a cell-mediated and humoral immune response. 74,82,109 Mice that are transgenic for MOG-specific T-and B-cell receptors develop spontaneous EAE. 14,60 Moreover, it has been shown that autoantibodies against MOG opsonize myelin and elicit demyelination via cytotoxicity of complement and macrophages. 67 However, not all detected anti-MOG antibodies seem to be capable of inducing the disease; antibodies against conformational and glycosylated MOG can mediate demyelination in EAE, whereas antibodies against linear MOG antigen that lack the correct membrane topology and show aberrant glycosylation are not able to induce demyelination. 72,119
The Role of Adjuvants in the Severity and Incidence of EAE
Injected adjuvants, especially CFA and IFA, are important elements for EAE induction and modulate incubation period as well as the overall severity of the disease. CFA is composed of paraffin oil and extracts of Mycobacterium tuberculosis, which stimulates an indiscriminate immune response in vivo, especially skewing T-cell differentiation toward the TH1 pathway. 15 In addition, this adjuvant produces a systemic inflammatory response inducing the subsequent opening of the BBB. 80 For these 2 reasons, the use of CFA proved extremely efficient in inducing an adaptive response to brain antigens.
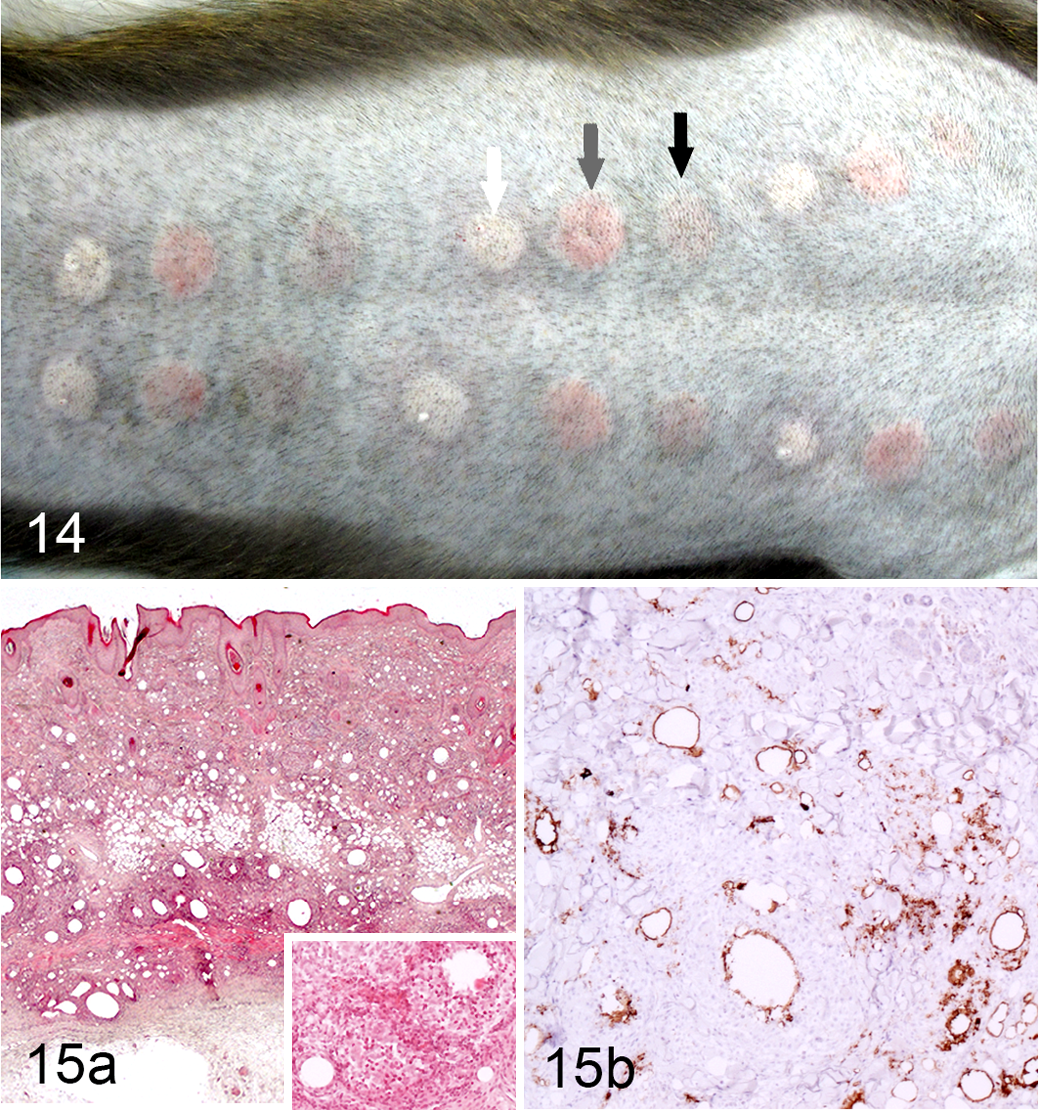
EAE in marmosets was first developed by replicating protocols developed for mice, which includes dermal immunization with human myelin formulated with CFA in combination with intravenous injection of Bordetella pertussis toxin. 73 In these conditions, animals develop an acute disease with severe necrotizing encephalitis. 34,104 Omission of the B. pertussis injection as well as decreasing the quantity of mycobacterial component in the CFA induces a less severe EAE, eventually with a remitting-relapsing clinical course. 104 In cynomolgus and rhesus macaques, myelin antigens formulated with CFA produce an acute and severe neurological disease associated with severe ulcerative skin granulomas formed at the immunization sites. 38 This substantial side effect makes CFA ethically unsuitable, especially for use in NHP. For this reason and to obtain a less aggressive form of the disease, IFA was chosen to induce EAE in macaques and the common marmoset. The use of IFA combined with rhMOG was shown to induce milder and eventually more chronic forms of EAE in marmosets and cynomolgus macaques. In contrast, in rhesus macaques immunized with rhMOG/IFA, the incidence of EAE was reduced (40%), and the severity of the disease remained similar as when using CFA as an adjuvant (Table 1). 57 Importantly, skin inflammation at rhMOG/IFA immunization sites was much weaker and transient than observed with CFA, showing a mild granulomatous dermatitis without ulceration (Fig. 14, 15). 38
The Immunopathogenesis of EAE
As mentioned, it is very likely that a specific autoimmune reaction against myelin causes IIDD in humans and in EAE. Several key steps of this cascade of events have been identified, tested, or confirmed in different EAE models. 22
Within the CNS, local APC and infiltrating neutrophils and macrophages are required for the development of tissue lesions, highlighting the importance of the innate immune system in this process. 38 Resident microglia present myelin antigens to the autoreactive T-cells, initiating the inflammatory reaction, and are responsible together with infiltrating macrophages and lymphocytes for the production of proinflammatory cytokines and chemokines (eg, IL1β, TNF-α, IFN-γ, MIP-1, CCL-2). 16,64 However, in the course of the inflammatory process, microglia (especially M2 microglia) might have multiple roles, including tissue repair and emission of anti-inflammatory signals (eg, IL-10 and TGF-β). 16,28,62 When these anti-inflammatory signals overwhelm the proinflammatory signals emitted by activated microglia/macrophages, the inflammatory activity of the lesion decreases, macrophages and neutrophils disappear, and remyelination begins, as seen in the marmoset EAE model with chronic lesions. 56 However, when anti-inflammatory signals are not able to restrain inflammation, the lesion enlarges, leading to the exacerbation of clinical signs and eventually death of the animal, as often observed in fulminant forms of the disease in rhesus and cynomolgus models. 38,39
Adaptive immunity plays a central role in EAE pathogenesis and especially during the earliest stages of lesion formation. As in rodents, the disease in NHP starts with CD4+ lymphocyte activation and their differentiation into encephalitogenic autoreactive cells. 26,38 This has been demonstrated many times in rodents and corroborated in NHP in experiments that either deplete CD4+ T-cells in cynomolgus macaques, prevent CD4+ T lymphocyte co-stimulation at priming in rhesus macaques, or prevent Th1/Th17 differentiation in marmosets. 17,37,94,105
Soluble pro-inflammatory factors produced by T- and B-cells, especially cytokines (eg, IL-6, IL-17), and anti-myelin antibodies are the second important factor in the development of inflammatory and demyelinating lesions. These soluble factors may be produced outside the brain and transported across a permeable BBB, but it seems more likely that they are produced locally. To support this hypothesis, the presence of T- and B-cells in early lesions indicates that at least part of these soluble pro-inflammatory factors are produced locally within the perivascular cuff, due to their central position within the lesions, and might be responsible for the lesion persistence and perpetuation of the myelin damage. In fact, in most EAE models, specific anti-myelin immunoglobulins represent one of the first measures of immune activation. Thus, it appears very likely that at the time of influx of macrophages and neutrophils into the lesion, activated complement and myelin-specific antibodies already coat myelin. The evidence that chronic lesions in the marmoset EAE model tend to increase in size even as BBB permeability decreases supports this interpretation. 71
Pathophysiologic Concepts of the Development of Human IIDD and EAE
So far, 2 main concepts currently dominate and are based on observations collected from studies of MS pathology as well as EAE modeling. 26
According to an “outside-in” paradigm, as in EAE, MS is caused by an event exogenous to the brain (eg, general viral infection), which in genetically susceptible individuals induces peripheral activation of autoreactive T- and B-cells. Lymphocytes with autoreactive potential against myelin antigens are present in MS patients as well as healthy individuals. 85 According to this hypothesis, although such autoreactive cells (principally CD4+ lymphocytes) are primed in the periphery, they differentiate into Th1 and Th17 CD4+ encephalitogenic lymphocytes that infiltrate the CNS and elicit inflammation following re-stimulation by local APC. 99 In opposition to this view and according to a so-called “inside-out” paradigm, a yet unidentified pathogenic event within the CNS, putatively infectious or degenerative, causes instability of myelin and release of myelin antigens, which induces activation of autoreactive T- and B-cells in CNS draining lymph nodes with further encephalitogenic properties. 99,107 Recently ‘t Hart and colleagues 107 proposed a unifying scheme of pathogenesis in which 3 compartments are considered critical for the development of EAE and MS (Fig. 16). According to this postulate based on studies in NHP EAE models and human clinical data, autoreactive T lymphocyte proliferation is actively induced by peripheral stimuli in EAE or MS. 45,106 Autoreactive T-cells then accumulate in lymph nodes and spleen (afferent compartment), followed by their migration to the CNS (target compartment), where they recognize their antigen on local APC, inducing their full activation and starting an inflammatory cascade leading to tissue injury. Tissue debris cleared from the CNS is often found in the macrophages within a third CNS-draining compartment, comprising the cervical and lumbar lymph nodes and the spleen. 107 Drained CNS antigens are presented to T-cells in this third compartment, which may lead to the generation of new T-cells autoreactive to novel brain antigens as well as epitope spreading. Such cells are released into the afferent compartment, where they can either mitigate or exacerbate the ongoing inflammatory reaction. 107 This 3-compartment model is supported by the presence of different myelin antigens (ie, MOG, PLP, CNP) in cervical lymph nodes and spleen in NHP EAE models (rhesus, cynomolgus, and common marmoset) as well as in MS patients. 24,116,117 Furthermore, the removal of cervical lymph nodes in remitting-relapsing mouse EAE model delayed the onset of relapses, underlining the importance of the CNS draining compartment in EAE pathology. 115
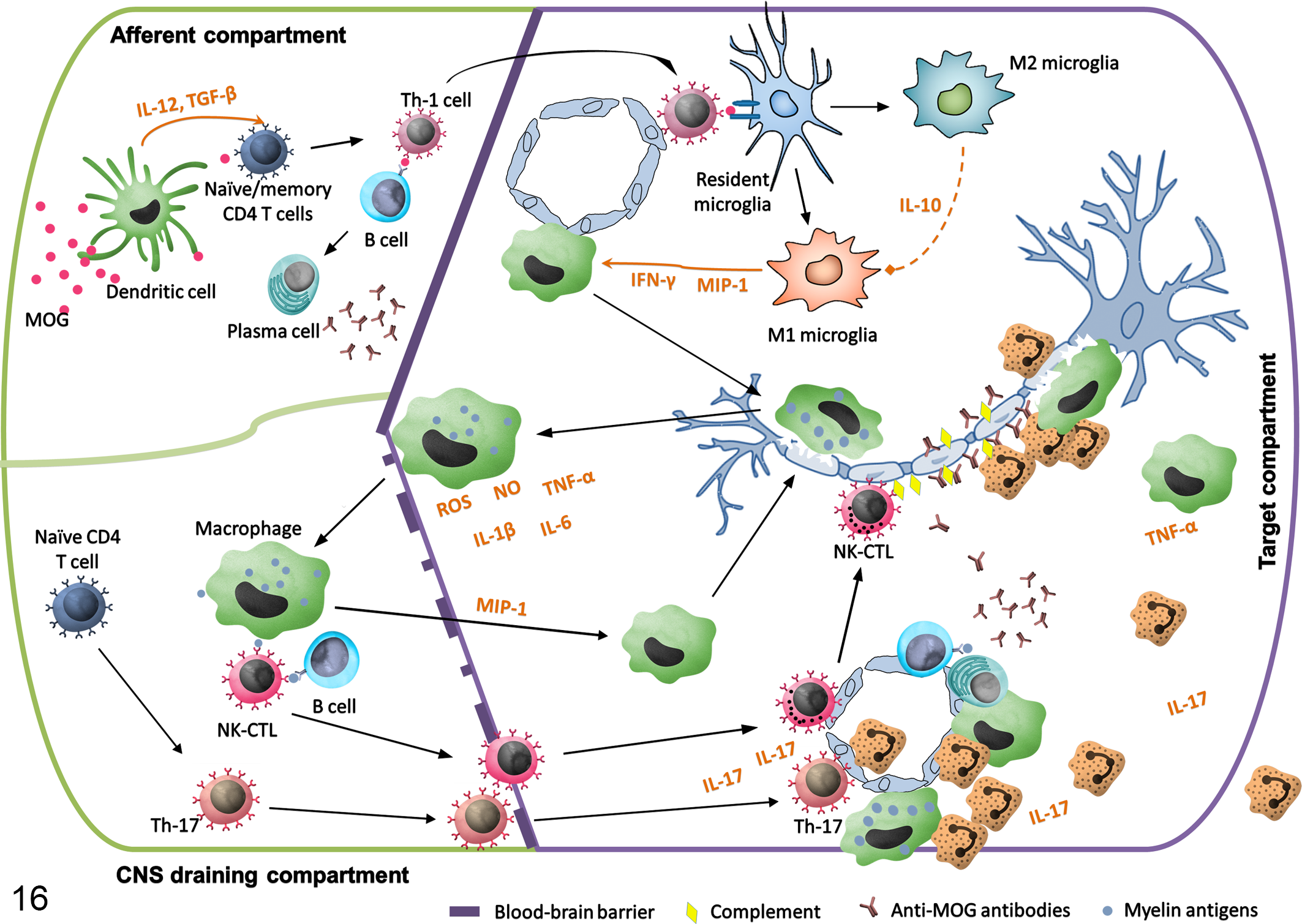
Immunopathological pathways implicated in the development of experimental autoimmune encephalomyelitis (EAE). In the afferent compartment (peripheral lymph nodes and spleen), injected myelin oligodendrocyte glycoprotein (MOG) antigen is presented by dendritic cells to naïve or autoreactive memory T- and B-cells and trigger a type 1 (TH1) immune response. TH1 cells migrate to the central nervous system (CNS) (target compartment), where they recognize their antigen on local antigen-presenting cells, become fully activated, and initiate an inflammatory cascade leading to the tissue injury. At the same time, M2 microglia start to produce anti-inflammatory signals (ie, IL-10, TGF-β), which tend to decrease the inflammation. Myelin debris that has been phagocytized by macrophages is evacuated to the CNS draining compartment (cervical and lumbar lymph nodes and spleen) where they elicit a novel activation of T- and B-cells and lead to the activation of the TH17 pathway. Activated T-cells (especially TH17 cells) and natural killer cytotoxic lymphocytes (NK-CTL) migrate through the permeabilized blood-brain-barrier together with numerous neutrophils and macrophages attracted by activated microglia. Epitope spreading occurs during myelin antigen presentation and anti-MOG conformational antibodies are formed. Myelin-specific antibodies, probably produced locally in the perivascular cuffs, together with complement, opsonize the myelin and attract numerous neutrophils and macrophages.
Pathophysiologic Pathways Implicated in the Development of EAE
Over the past few decades, EAE NHP models have made it possible to study the pathogenesis of inflammatory and demyelinating lesions in CNS. Studies in the common marmoset EAE model have particularly illuminated the fact that 2 pathways are important for the development of EAE: the initiation and the progression pathways. 109
The initiation pathway begins with peripheral presentation of myelin antigen bound to MHC-II by APC to the autoreactive T-cells, which permits development of encephalitogenic lymphocytes and TH1-driven inflammation in the white matter of the brain and spinal cord.
These lymphocytes initially migrate to the perivascular spaces via transendothelial diapedesis, the cerebrospinal fluid compartment, or the choroid plexus during the physiological process of immunological surveillance of the CNS. 43,68,84,90,122 T-cell activation is followed by chemokine and cytokine production, endothelial cell activation, increase of BBB permeability, and a subsequent influx of macrophages and neutrophils that degrade myelin. 71 The pathogenic relevance of the initiation pathway is illustrated by the treatment with anti-IL-12p40 monoclonal antibody (Ustekinumab) of marmosets immunized with MOG. Early treatment (before clinical signs) with Ustekinumab completely abrogates EAE, whereas if provided after disease onset, the efficacy is poor in inducing the suppression of inflammation and preventing enlargement of the lesions. 105 Thus, it was hypothesized that during the course of the disease, a second pathogenic mechanism opening a path toward the chronic condition must be activated. A possible initiator of this progression pathway might be the release of self-antigens during the initial inflammation of the brain. These antigens, especially MOG epitopes, would elicit additional CNS inflammation, leading to progressive myelin destruction (Fig. 16). 107
Different cell types are implicated in these 2 phases of the development of EAE. Fine mapping of the T-cell response in marmosets immunized with rhMOG revealed the specific activation of TH1 cells for the epitope MOG(24–36). These cells induce a rapid EAE onset during the first phase of the disease. 56 The initial inflammatory change is followed by a late-stage demyelination, which is driven by another T-cell fraction, specific for the epitope MOG(40–48). This T-cell subpopulation is CD3+CD4+CD8α+CD56+CD28-CD27+CD16-CCR7-, produces a high level of IL-17A, and holds a specific cytotoxicity against B-cells transformed with EBV and presenting peptides derived from MOG(40–48). 49,56,109 Moreover, such a cell population is very similar to a subset of natural killer (NK) cell-related cytotoxic T-cell lymphocytes found in humans and named natural killer cytotoxic T lymphocytes (NK-CTL). 75,108 These pathogenic T cells are specific for MOG(40–48) and to the presenting molecule MHC Caja-E (similar to HLA-E in humans). Furthermore, in this model, due to the absence of anti-MOG IgG and IgM antibodies, which is a usual requirement for the capacity of NK-CTL to mediate demyelination, it was concluded that the autoreactive NK-CTL are directly responsible for demyelination. 49 The specific epitopes MOG(24–36) driving the TH1 initiation pathway and MOG(40–48) driving the NK-CTL mediated progression pathway are positioned in the highly conserved portion (20–50) of the MOG extracellular domain. There is only 1 difference in the position 42, which is occupied by a serine in mouse, rat, and marmoset or a proline in human and macaque. 108
Infectious Influences in EAE Development
Similar to humans, viral and microbial infection is in our opinion the most important infectious factor influencing the severity of EAE in NHP models.
Spontaneous encephalomyelitis in some animals has been described with MS-like features, such as virus-induced demyelinating disease in Japanese macaques. 4,97,102 Indeed, animals infected with Japanese macaque rhadinovirus (JMRV) showed clinical, MRI, and pathological features reminiscent of ADEM but also MS. The CNS of infected monkeys contained multifocal demyelinating lesions of varying ages, including acute active demyelinating lesions with macrophages and lymphocytic periventricular infiltrates as well as chronic, inactive demyelinated lesions with low inflammatory changes. 4 However, given the infectious nature of this disease, the link to the pathogenesis of MS and other IIDD is unclear. Another spontaneous infectious demyelinating disease in macaques was described in immunocompromised animals. Simian virus 40 (SV40), family Polyomaviridae, can cause a fatal demyelinating CNS disease in immunocompromised macaques analogous to progressive multifocal leukoencephalopathy caused by John Cunningham (JC) virus in immunocompromised humans. 54 SV40 infects mainly neurons, astrocytes, and oligodendrocytes in cerebrum and cerebellum and more rarely in the spinal cord, inducing multifocal necrosis and perivascular lymphoplasmacytic cuffing, mainly in subpial white matter and at the white-grey matter junctions. In addition, as in humans with JC virus-induced disease, macaques can develop SV40-associated CNS demyelination especially in the subpial white matter. 5
In addition, concomitant seasonal bacterial or viral infections (eg, upper respiratory tract infections) may influence EAE severity or shorten the time to EAE onset. We observed a coincidence of seasonal infections during the winter months (including viral rhinitis) with more rapid onset of EAE in cynomolgus macaques, underlining the importance of viral infections in the context of EAE development (unpublished results).
Concluding Remarks
Currently available NHP EAE models recapitulate at least part of the spectrum of IIDD in humans. Whether acute (ie, ADEM, clinically isolated syndrome [CIS], or NMO) and chronic demyelinating diseases (ie, MS) have a common original pathogenesis is still debated, as is whether EAE recaps ongoing pathological processes of either or all of these diseases. 44,106 In any case, NHP EAE models present a similar clinical and pathological appearance to acute human IIDD, with ADEM-like inflammatory changes in rhesus and cynomolgus macaques immunized with myelin antigen in CFA or IFA. In some cases, these models mimic chronic relapsing-remitting demyelinating lesions resembling MS, as in common marmoset or some cynomolgus individuals immunized with rhMOG/IFA. Thus, EAE in NHP is a heterogeneous disease, which makes it a very versatile system to use in translational neuropathological and immunopathological research. The disease variability across species and immunization protocols also creates an opportunity to explore multiple facets of the immune-mediated mechanisms of neuro-inflammation and demyelination, as well as intrinsic protective mechanisms, and develop more effective and safe therapies.
Footnotes
Acknowledgements
We wish to thank Anne-Laure Bauchet for providing macroscopic and microscopic photographs, Sophie Luccantoni for excellent technical assistance, and Susannah Williams for correction of this manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.