
Abstract
Clinical, gross, histopathologic, electron microscopic findings and enzymatic analysis of 4 captive, juvenile springboks (Antidorcas marsupialis) showing both polycystic kidneys and a storage disease are described. Springbok offspring (4 of 34; 12%) were affected by either one or both disorders in a German zoo within a period of 5 years (2008–2013). Macroscopic findings included bilaterally severely enlarged kidneys displaying numerous cysts in 4 animals and superior brachygnathism in 2 animals. Histopathologically, kidneys of 4 animals displayed cystic dilation of the renal tubules. In addition, abundant cytoplasmic vacuoles with a diameter ranging from 2 to 10 μm in neurons of the central and peripheral nervous system, hepatocytes, thyroid follicular epithelial cells, pancreatic islets of Langerhans and renal tubular cells were found in 2 springbok neonates indicative of an additional storage disease. Ultrastructurally, round electron-lucent vacuoles, up to 4 μm in diameter, were present in neurons. Enzymatic analysis of liver and kidney tissue of 1 affected springbok revealed a reduced activity of total hexosaminidase (Hex) with relatively increased HexA activity at the same level of total Hex, suggesting a hexosaminidase defect. Pedigree analysis suggested a monogenic autosomal recessive inheritance for both diseases. In summary, related springboks showed 2 different changes resembling both polycystic kidney and a GM2 gangliosidosis similar to the human Sandhoff disease. Whether the simultaneous occurrence of these 2 entities represents an incidental finding or has a genetic link needs to be investigated in future studies.
Polycystic kidney disease (PKD) is characterized by multiple cysts in 1 or both kidneys and other organs such as liver and pancreas in humans and animals. 15,38,60 Various mutations in genes regulating development and function of renal tubular epithelial cells have been detected as causes of PKD in various species. 60 Different gene defects in PKD lead to protein dysfunction, resulting in dedifferentiation of cells, excessive fluid secretion, and proliferation of tubular epithelia causing renal cysts. 57 PKD with an autosomal dominant and autosomal recessive mode of inheritance due to mutations in different genes has been described. 57 In humans, autosomal dominant PKD may occur due to mutations in the PKD1 and PKD2 genes affecting proteins, which play an important role in the physiology of renal tubular cells, primary cilia, and basal bodies. 57 Human autosomal recessive PKD causes cysts in kidneys and the liver and is associated with defects in the PKHD1 gene. 4,53 PKD is also described in many domestic animals, including cats and dogs. 10,37,60 In cats, a stop mutation with a C>A transversion was identified in the PKD1 gene, probably leading to a deficiency in the respective enzyme. 32,62 Persian and Persian-related cats are overrepresented breeds with PKD. 5,10 Furthermore, renal cysts of unknown causes or due to environmental pollutants have been described in several species, such as goldfish (Carassius auratus), hippopotamus (Hexaprotodon liberiensis), white-tailed deer (Odocoileus virginianus), European roe deer (Capreolus capreolus), red panda (Ailurus fulgens), and Northern pintail (Anas acuta). 6,33,42,43,48,54 In addition, cystic kidneys can also be caused experimentally (eg, steroids in hamsters or obstructions due to prominent scar tissue). 8,12,36,58 Pathogenetically, administration of adrenocortical steroids in newborn hamsters may contribute to the development of cystic tubuli by a potassium depletion and a drug-induced reduced elasticity of the tubular basement membrane for unknown reasons. 12
In animals, lysosomal storage diseases usually affect multiple organs and primarily cells with an increased metabolism. 25 The stored material is usually deposited in the cytoplasm of defective cells, resulting in a severe vacuolation with loss of function. In veterinary medicine, nearly all storage diseases are related to lysosomal defects and are either inherited or acquired. 20,44 Inherited lysosomal storage diseases show deficient enzymatic function of specific enzymes due to a genetic defect. 25 Storage of the material commonly starts in fetal tissues during pregnancy and becomes obvious at birth or in the weeks thereafter. 36,40,41 Only few diseases develop clinically prominent signs with advanced age. Lysosomal storage diseases can be classified based on the underlying molecular defect in primary lysosomal hydrolase defects, faulty posttranslational processing of lysosomal enzymes, disturbed trafficking lysosomal enzymes, lack of lysosomal enzyme protection, or deficiencies of soluble nonenzymatic lysosomal proteins. 24 Furthermore, lysosomal storage diseases can also be distinguished according to the accumulated product (eg, sphingolipids, glycolipids, oligosaccharides, or mucopolysaccharides). 25 Neuronal storage diseases occurring in animals include sphingolipidoses, such as GM1 and GM2 gangliosidosis; mucopolysaccaridoses; and glycoproteinoses, such as mannosidosis. 11,28,39
Sphingolipidoses belong to a group of inherited storage diseases characterized by a defective degradation of glycosphingolipids. 25 These diseases cause storage of sphingolipids in neuronal lysosomes. 36 The enzyme β-galactosidase in GM1 gangliosidosis and hexosaminidase (Hex) in GM2 gangliosidosis are defective, causing a reduced enzymatic activity or its absence. 2,29,35,51,55 GM2 gangliosidosis can occur in 3 different variants: the 0-, B- and AB-variant. The 0-variant represents the Sandhoff (-like) disease characterized by a deficiency in the HexA and HexB due to a HEXB gene mutation in the β-subunit. 18,26,51 The B-variant displays a HEXA gene mutation in the α-subunit with a defective HexA, leading to Tay-Sachs disease. 56 In the AB-variant, HEXA and HEXB genes are normal, and the mutation occurs in the GM2A-gene, causing a lack of GM2 function. 34 In animals, all 3 variants of GM2 gangliosidosis have been described. A deficiency of the lysosomal β-hexosaminidase A and B, which is typical for a 0-variant resulting in Sandhoff (-like) disease, has been described in a Golden Retriever dog, Toy Poodles, pigs, Japanese domestic cats, Korat cats, and European Burmese cats. 7,26,27,45,51,61 In Jacob sheep and the Muntjak deer, a defect in the HexA was found representing the classic B-variant of GM2 gangliosidosis. 13,56 In cats and other species, a mutation of the GM2 activator protein (AB-variant) was defined, causing severe motor disturbances. 34
To shed light on an unusually high number of postnatal losses in a herd of captive springboks, the aims of the present study were 4-fold: (1) a detailed description of the pathologic findings in 4 related animals, (2) a pedigree analysis of affected animals, (3) a comparative enzymatic analysis of liver and kidney, and (4) a comparison to previously described similar diseases in this species.
Materials and Methods
Animals
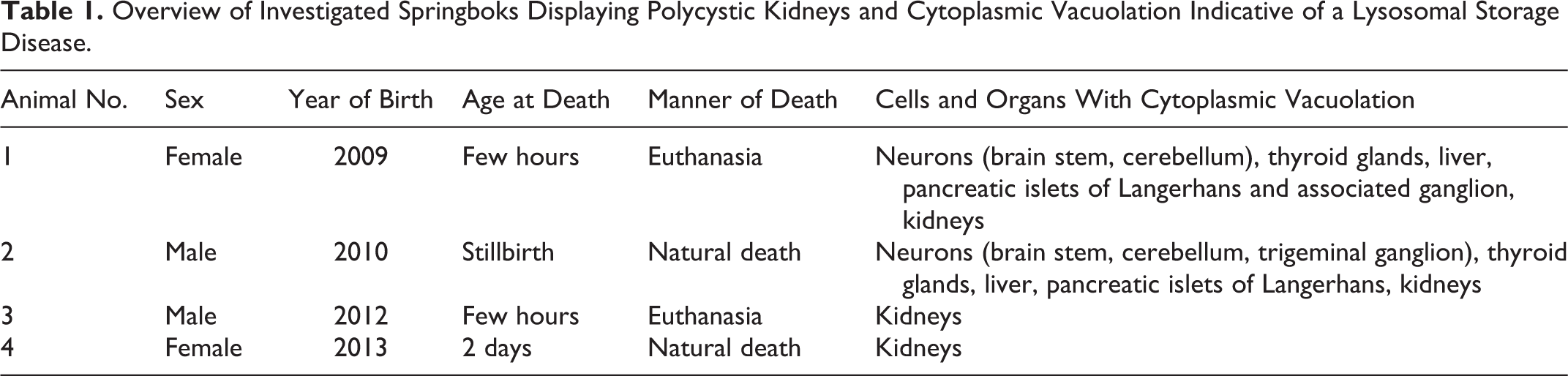
A total of 34 springboks were born at Hannover Zoo, Lower Saxony, Germany, between 2008 and 2013. Twelve of 26 animals died naturally, 13 animals were euthanized, and the cause of death in 1 animal remained undetermined. Twenty-five of 26 animals were submitted for necropsy. Four of these springboks showed polycystic kidneys, and these 4 animals (animal Nos. 1–4) are in the focus of the study. Although Raboisson et al 50 defined the neonatal period from 3 days to 1 month of age in calves, in the present study, neonates also included animals up to 2 months of age. This study was conducted in accordance with the German Animal Welfare Act. All animals were submitted dead for necropsy to investigate the cause of stillbirth or diseases. The urea concentration in the anterior eye chamber was measured to evaluate renal function. Postmortem urea concentration in the aqueous humor correlates well with the ante mortem urea concentration in the blood and aqueous humor. 19 The authors confirm that no animals were sacrificed for the purpose of this study. Table 1 summarizes signalment and the most important findings of the 4 affected springboks.
Overview of Investigated Springboks Displaying Polycystic Kidneys and Cytoplasmic Vacuolation Indicative of a Lysosomal Storage Disease.
Light and Transmission Electron Microscopy
Tissue samples were taken during necropsy and fixed in formalin, routinely embedded in paraffin wax, cut at a 3-μm thickness, and stained with hematoxylin and eosin (HE). The following tissues were taken for histology from affected animals (animal Nos. 1–4): bone marrow, lung, spleen, liver, kidneys, thyroid glands, adrenal glands, skeletal muscle, myocardium, pancreas (with associated ganglion in animal No. 1), abomasum, omasum, reticulum, rumen, small and large intestine, cerebrum, cerebellum, and brain stem. Spinal cord, umbilicus, nose, tongue, thymus and pituitary gland, rete mirabile (animal Nos. 2–4), central and peripheral nerves (sciatic nerve; animal Nos. 2–4), optic nerve (animal No. 2), trigeminal nerve with ganglion (animal Nos. 2–4), nerves of plexus brachialis (animal Nos. 2, 3), testes (animal Nos. 2, 3), uterus and ovaries (animal No. 4), urinary bladder (animal No. 2), eyes (animal Nos. 2, 4), salivary glands, and mesenteric lymph nodes (animal No. 3). Brain stem and cerebellum also were stained with Luxol-fast blue (LFB), Bielschowsky’s silver impregnation, and periodic acid–Schiff (PAS) reaction as described. 52 Electron microscopic pop-off technique using brain stem (animal Nos. 1, 2), trigeminal ganglion, liver (animal No. 1), and kidney (animal Nos. 2, 4) was performed as previously described. In addition, scanning electron microscopy was performed as previously described. 3,21,46
Pedigree Analysis
The pedigree of affected animals was investigated to reveal the mode of inheritance of the observed findings.
Enzymatic Analysis
For enzymatic analyses, fresh, snap-frozen liver and kidney tissues of 5 springboks (animal No. 2 and 4 nonaffected springboks of various ages; Tables 1 –3) were taken during necropsy and stored at –80°C until use. Tissues were processed according to Torres et al. 56 Briefly, samples (approximately 1 g) were homogenized in distilled water (10% w/v) and centrifuged at 30 000 g for 20 minutes at +4°C. Protein concentration was determined according to Lowry et al. 31 The supernatants were used to determine the activity of selected lysosomal enzymes using commercially available fluorescent (4-methylumbelliferyl; 4-MU) substrates. α-Mannosidase, α-galactosidase, and α-fucosidase were assayed according to Gehler et al. 17 β-Mannosidase was assayed according to Pearce et al 49 and total hexosaminidase and hexosaminidase A according to Torres et al. 56
Hepatic Enzyme Activities of Affected Springbok No. 2 and 4 Control Animals.
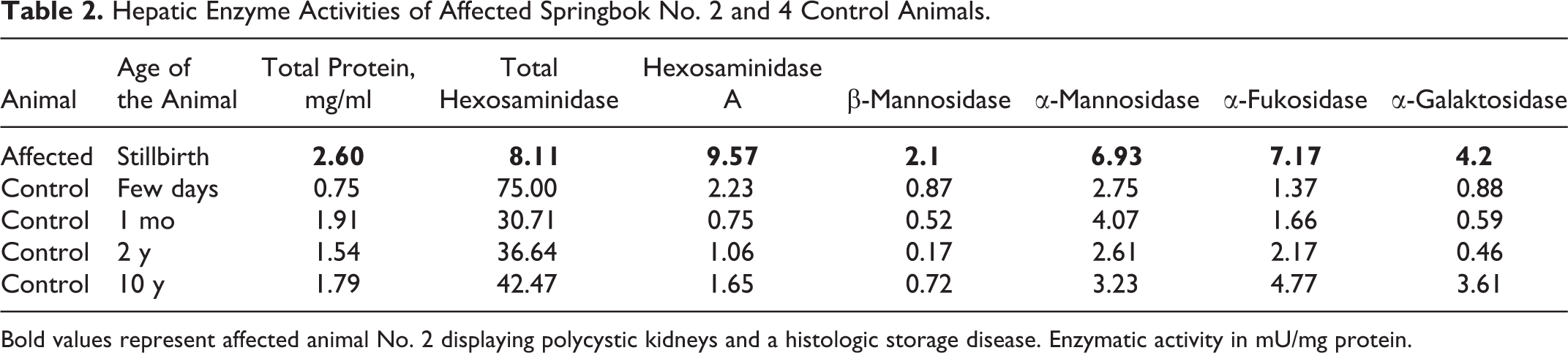
Bold values represent affected animal No. 2 displaying polycystic kidneys and a histologic storage disease. Enzymatic activity in mU/mg protein.
Renal Enzyme Activities of Affected Springbok No. 2 and 4 Control Animals.
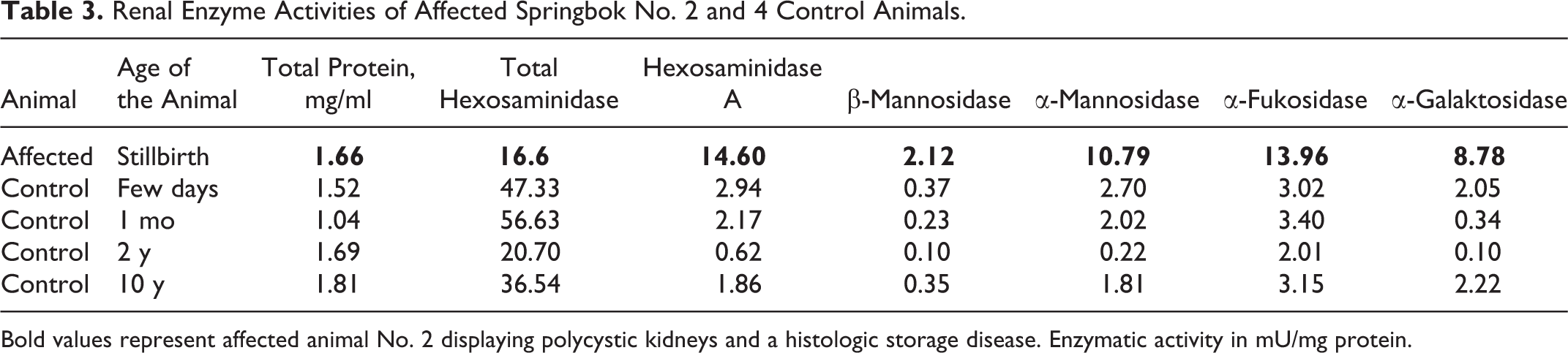
Bold values represent affected animal No. 2 displaying polycystic kidneys and a histologic storage disease. Enzymatic activity in mU/mg protein.
Results
Animals and Clinical Findings
Within a 5-year period (2008–2013), 34 springboks were born at Hannover Zoo. Twenty-six of these animals (26/34 = 76.5%) died during this time. Age, sex, and manner of death of 4 of these springboks (animal Nos. 1–4) are listed in Table 1. The remaining 22 animals included 8 male and 14 female springboks. Eleven of these 22 animals were 2 months old or younger. They are termed neonates for clarity throughout this article. The remaining 11 animals were between 10 months and 11 years old. Animals lived from a few hours to 15 years, and they died naturally or were euthanized. Cause of death of the neonates included inadequate colostrum uptake with suspected hypoglycemia-hypothermia (4 cases); purulent omphalophlebitis, meningitis, or polyarthritis (1 case of each); trauma with a fracture of a leg (1 animal); and no specific diagnosed disease (3 cases). Only 1 animal was not submitted for necropsy.
Macroscopic Findings
Four (animal Nos. 1–4) of 15 (27%) neonatal springboks displayed a severe, diffuse enlargement of both kidneys (renomegaly) with numerous large cysts, up to 0.5 mm in diameter (Figs. 1, 2). In all 4 cases, the ureter and urethra were normal. Additional findings included anemia consisting of pale skeletal musculature and a severe, acute, retroperitoneal and extracapsular hemorrhage of the left kidney in animal No. 3 (Fig. 1). Animal Nos. 1 and 3 displayed a superior brachygnathism, complete fetal atelectasis, and aspiration of amniotic fluid was present in animal No. 2. The brain of the 4 affected animals showed no significant macroscopic findings.

Springbok, neuronal storage disease and polycystic kidneys.
Aqueous Urea Concentrations
Animal Nos. 3 and 4 showed an increased amount of urea in the anterior eye chamber. Only animal No. 4 displayed severe azotemia (urea concentration in the anterior eye chamber: 240 mg/dl); the value of animal No. 3 was only slightly increased (mild azotemia; 70 mg/dl). Animal Nos. 1 and 2 showed urea concentrations ≤50 mg/dl (reference value: ≤50 mg/dl).
Light Microscopy
The kidneys of animal Nos. 3 and 4 were characterized by diffusely distributed and severely enlarged cystic tubules (80% of all tubules) lined by a flattened or rarely cuboidal epithelium of the renal medulla and cortex (Fig. 3). Infrequently tubular cysts displayed luminal projections with flattened epithelium. In addition, multifocally few lining epithelial cells of dilated tubules displayed a polypoid hyperplasia. Between severely dilated tubules, few nondilated tubules exhibited cytoplasmic vacuoles, approximately 5 μm in diameter. Moderate tubular cyst formation was noted in the kidneys of animal Nos. 1 and 2. In these 2 animals, cystic tubules were located primarily in the renal medulla, presumably affecting the distal part of the nephron, while cortical areas were only rarely affected. In addition, the tubulointerstitial fibrovascular tissue was prominent and the urinary space of multiple glomeruli was slightly to moderately dilated in all 4 animals. Neurons in various regions of the central and peripheral nervous system, including brain stem, cerebellum, trigeminal ganglion (animal No. 2), and peripheral ganglion (animal No. 1) displayed abundant cytoplasmic vacuoles with a diameter ranging from 2 to 10 μm (Fig. 4), as detected in HE-stained sections. In the cerebellum, Purkinje cells were predominantly affected. Neuronal vacuoles were neither marked by Bielschowsky’s silver impregnation (Fig. 5) nor PAS reaction. However, the center of the neuronal vacuoles was stained blue by LFB staining (Fig. 6). Animal Nos. 3 and 4 did not show any neuronal changes. Thyroid follicular epithelial cells (Fig. 7), hepatocytes (Fig. 8), and pancreatic islets of Langerhans of animal Nos. 1 and 2 also displayed a moderate to severe cytoplasmic vacuolation. Cytoplasmic vacuolation of islets of Langerhans was substantiated by a synaptophysin immunohistochemistry (data not shown).
Springbok, neuronal storage disease.
None of the springbok neonates (animal Nos. 1–4) displayed cysts in the pancreas and the liver. Kidneys of animal Nos. 1 and 2 showed a vacuolation of epithelial cells in nearly all tubules in the renal medulla and cortex (Fig. 9). In animal Nos. 1 and 2, all tubules of the nephron were equally affected by vacuole formation. There was no predilection of proximal convoluted tubules as previously described. 23 However, animal Nos. 3 and 4 lacked widespread tubular vacuolation. In these animals, the kidneys only occasionally displayed slightly vacuolated tubular epithelial cells.
Electron Microscopy
Hepatocytes, renal epithelial cells, and neurons (Fig. 10) contained numerous variably sized, partly coalescing, electron-lucent vacuoles within their cytoplasm. Some vacuoles were surrounded by membranes and filled with variable amounts of a moderate electron-dense material. In addition, the majority of investigated renal tubular epithelial cells displayed an intact, homogeneous basement membrane, a continuous brush border, and few electron-dense lysosomal bodies (Fig. 11).
A scanning electron microscopic investigation revealed multifocally mild protrusions of the apical plasma membrane originating from renal tubular epithelium leaping into the luminal space (Fig. 12).
Enzymatic Analysis
The assay of selected lysosomal enzymes in liver and kidney tissue homogenates of animal No. 2 revealed increased activities of β-mannosidase, α-mannosidase, α-fucosidase, and α-galactosidase compared with unaffected control animals, thus ruling out β-mannosidosis, α-mannosidosis, fucosidosis, and Fabry disease (α-galactosidase deficiency). The activity of total hexosaminidase, however, was significantly reduced in both liver and kidney samples of this animal compared with the unaffected control animals. Furthermore, the activity of hexosaminidase A was increased, reaching the level of total hexosaminidase activity (Tables 2, 3). These findings suggested the likelihood of a hexosaminidase defect indicative of a GM2 gangliosidosis. Unfortunately, it was not possible due to the limited amount of available tissue to perform further biochemical analyses to elucidate the exact enzyme defect. However, it would have been valuable to investigate the activity of HexB to find out more about the underlying enzyme defect.
Pedigree
Closely related male and female animals were affected equally. The affected animals were born from nonaffected parents. Although caution has to be applied due to the small numbers, the pedigree is compatible with a monogenic autosomal recessive mode of inheritance (Fig. 13).
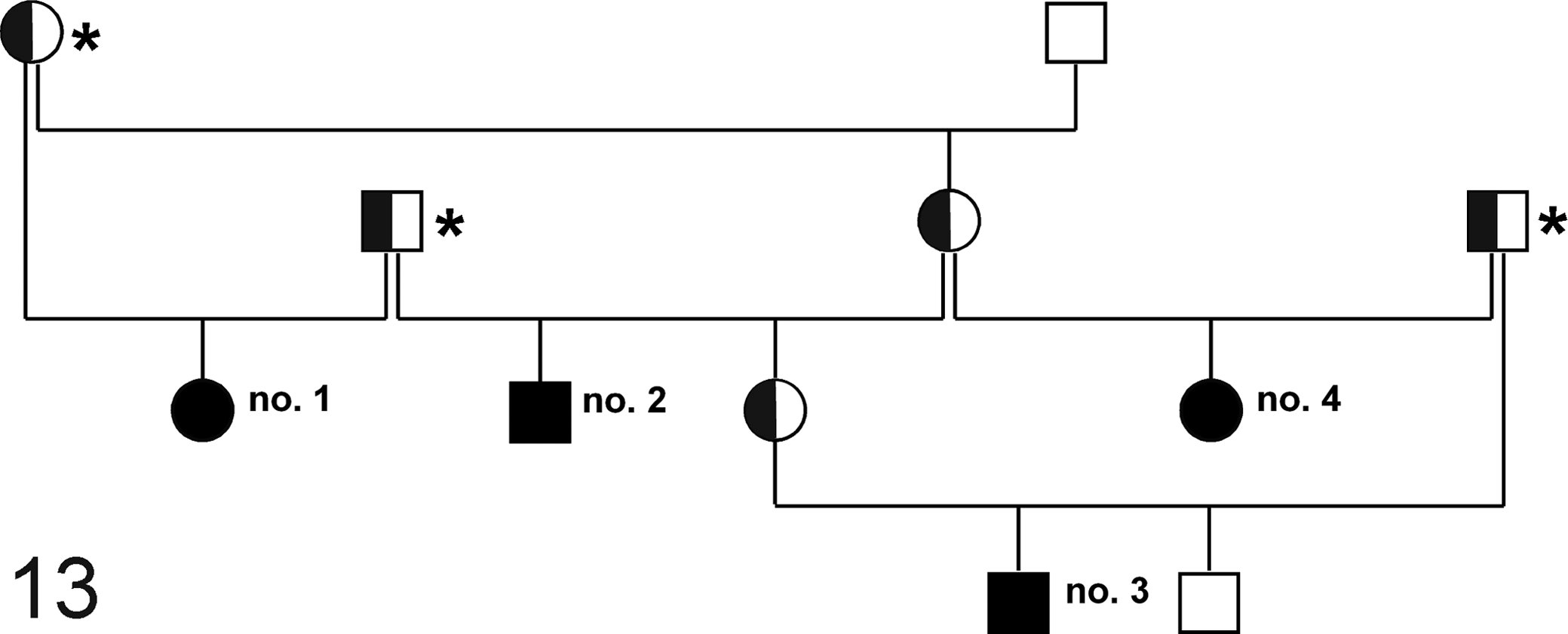
Pedigree analysis of springboks with polycystic kidneys and neuronal storage disease. Squares depict males and circles females. Filled and open symbols represent affected and nonaffected animals, respectively. Half-filled symbols represent obligate carriers. Animal Nos. 1 to 4 displayed polycystic kidneys and animal Nos. 1 and 2 also exhibited changes suggestive of GM2 gangliosidosis. All 4 affected animals (animal Nos. 1–4) can be traced back to 3 animals (*), which have common ancestors in France.
Discussion
The present study describes clinical, gross, histopathologic, enzymatic, and electron microscopic findings in 4 related springbok neonates displaying 2 different most likely inherited diseases with polycystic kidneys and a storage disease, which is similar to a GM2 gangliosidosis with a defective hexosaminidase. In addition, a pedigree analysis of the affected animals is provided. The 4 springboks were stillborn, died, or were euthanized within the first 2 days after birth because of developmental and metabolic disturbances. Although there was no disease-specific clinical signs for either entity, severe azotemia was a prominent finding in 1 springbok neonate with cystic kidneys. In 2 cases, the renal pathology and storage disease were associated with superior brachygnathism.
Gross and histopathologic renal changes of affected springboks were characterized by polycystic kidneys with cystic tubules and flattened epithelium in both kidneys, similar to the morphology of 2 previous descriptions from France and the United States in the same species. 23,30 In the present study, renal cysts were not associated with cysts in other organs, in contrast to 1 previous study. 23 In cats and humans, PKD is associated with cysts in other organs such as exocrine pancreas and liver. 10,37,47,60 However, cystic changes were not observed in the liver or pancreas of in the springboks in the present study. 10,47 Electron microscopic findings of the kidney partly resembled changes described for autosomal recessive PKD in mice. 16 Similarly, cellular proliferations were detected at the apical surface of affected tubules by scanning electron microscopy. However, changes in the basement membrane such as thickening and fibrillation together with swollen microvilli and focal cytoplasmic extensions into the microvilli were absent. 16 In summary, ultrastructural analyses lacked final confirmation that changes resembled lesions described for PKD. In addition, light microscopy of the liver and pancreas lacked changes as described for PKD. Therefore, a possible genetic basis for the observed polycystic kidneys needs to be substantiated in further studies. Alternatively, dysplastic renal changes, acquired renal cysts, or cystic kidneys due to the accumulation of storage material in renal epithelial cells or other pathogenetic mechanisms must be considered. In addition to the polycystic kidneys, 2 springboks showed cytoplasmic vacuolation in neurons of the central and peripheral nervous system, thyroid follicular epithelial cells, pancreatic islets of Langerhans, hepatocytes, and renal tubular epithelial cells. The histopathology of all 4 animals and enzyme analysis of 1 affected springbok exhibiting cytoplasmic vacuolation suggest the occurrence of a lysosomal storage disorder with a presumed defect in the HEXB gene. This assumption is based on the reduction in the total hexosaminidase activity in the liver and kidney and a compensatory increase in the activities of other lysosomal enzymes. A previous description of a lysosomal storage disease in neonatal springboks showing similar tissue changes concluded that changes were the result of a mannosidosis. However, this assumption was solely based on morphological findings. 30 The fact that α- and β-mannosidase activities were not reduced in freshly obtained kidney and liver samples in one of the affected springboks compared with control animals provided no further support for such a conclusion. It is intriguing to speculate that these previously described animals had the same defect as the animals in the present study. Furthermore, the findings of the present study, especially the results of the enzyme assays, suggest a defect in the hexosaminidase genes, favoring a GM2 gangliosidosis of the 0-variant, also termed Sandhoff disease in humans. The reduction in the total hexosaminidase with an increased amount of HexA indicates a defective HexB, although it was not possible to measure the enzymatic activity of the HexB. Unfortunately, we were unable to further characterize the individual hexosaminidases (eg, by electrophoresis) 56 because of the limited amount of available frozen tissue. Therefore, a deficiency of a yet unknown hexosaminidase in springbok tissues cannot be ruled out. In addition, it was not possible to characterize the lipids present in neuronal tissue, which would add weight to the suspected diagnosis of a GM2 gangliosidosis. 55 The enzymatic analysis of control animals included tissue of adult and neonatal springboks. The activities of some enzymes, including renal and hepatic hexosaminidase (total and HexA), seemed to be lower in adult animals. Whether this observation indicates a general age dependency of enzyme activity needs to be substantiated in future studies.
Experimentally or spontaneously induced intoxications can cause similar cytoplasmic vacuolation in hepatocytes, thyroid gland epithelium, and Purkinje cells and/or cystic kidneys. 1,8,12,14 However, there was no indication of a toxic etiology of the described storage disease, such as, plant intoxication with Malvaceae (Sidacarpinifolia) and Convolvulaceae (Ipomoea carnea subsp fistulosa) as reported in cattle and goats. 1,9,14 However, to substantiate the suspected GM2 gangliosidosis of the 0-variant, possible mutations have to be determined by molecular and further enzymatic assays to confirm a hexosaminidase defect. In addition, an intoxication as the cause for the described storage disease and polycystic kidneys cannot be ruled out completely and warrant further studies.
In a previous publication, gross findings of springboks with storage disease included arthrogryposis and domed skull with a depressed nasal bridge. 30 However, in the present study, animals lacked arthrogryposis and other reported changes. Although such findings were not observed in the present study, similar findings may occur in a variety of congenital conditions with central nervous system involvement. 22,59
The gross pathology of the kidney, characterized by fluid-filled cysts and renomegaly, was reminiscent of 2 previously described cases of neonatal springboks from animal parks in France and the United States. 23,30 In addition, histopathologic, neuronal, and renal changes were very similar. 23,30
Information from the breeding data of the German zoo pointed toward obligate carriers in the pedigree analysis, which originated from an animal park in France, where the diseases had previously occurred. 30 Further pedigree analysis revealed that inbreeding could be a factor for the occurrence of both diseases. This hypothesis gains further support from the fact that all obligate carriers (and thus all affected animals) might have ancestors in 1 French animal park. However, this remains speculative since no specific records were available to confirm the assumption that polycystic kidneys and the neuronal storage disease originate from a single animal. Nonetheless, both the polycystic kidneys and the storage disease presumably followed a monogenic and autosomal recessive trait of inheritance.
Unfortunately, it was also not possible to link the pedigree of described animals in this study to the cases in the United States in the 1970s. 23 However, interestingly, the breeding facility in the United States included 2 dams from Germany born in 1965. It cannot be ruled out that this exchange of animals links the described cases. However, a relation can only be assumed and remains speculative. As previously described, PKD in springboks affected 18% of a herd within a 5-year period and occurred in both sexes. 23 These data, in combination with our findings, support the hypothesis that the disease is inherited and probably follows an autosomal recessive trait of inheritance. Furthermore, based on our findings, a monogenic trait is suggested. In the German animal park, 12% (4 of 34 born animals) of the springbok neonates showed cystic kidneys in the previous 5 years (2008–2013), indicating a similar frequency. Whether renal failure due to polycystic kidneys contributes significantly to neonatal death in captive springbok populations in Germany, France, and the United States needs to be investigated in future studies. 23,30
In summary, results of the present and previous studies suggest the concurrent occurrence of polycystic kidneys and a lysosomal storage disease, presumably a GM2 gangliosidosis, in German, French, and North American captive springbok populations. 23,30 Pedigree analysis indicates an autosomal recessive trait for both diseases and identifies obligate carrier animals, which originated from a French animal park. In addition, the present study also shows that affected springboks from previous descriptions seemed to be related to the affected population described here, and therefore, it is assumed that the present cases originated from the genetic pool in France and Germany. Further studies, however, are needed to elucidate the molecular basis of both diseases. Conclusively, inherited diseases must be considered a significant cause of stillbirth and neonatal death in captive springboks and might represent a major threat for this species.
Footnotes
Acknowledgements
The help of Robert Kreutzer, PhD, at the beginning of this work is gratefully acknowledged. We thank Bettina Buck, Petra Grünig, Kerstin Rohn, and Claudia Herrmann for their excellent technical assistance. We also thank Mrs Sherwood-Brock for proofreading the manuscript.
Authors’ Note
The contents of this article were orally presented in Fulda, Germany at the “57. Jahrestagung der Fachgruppe Pathologie der Deutschen Veterinärmedizinischen Gesellschaft” (8–9 March 2014), and an abstract was published in Tierärztliche Praxis 2014;2:A9.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.