
Abstract
Coagulation factor XII (FXII) may be important in cardiovascular and inflammatory diseases. We have identified and characterized a naturally occurring mutation in the feline FXII gene that results in a mutant protein and enzymatic loss of activity. Feline intron/exon gene structure and sequence were acquired by comparing DNA sequences obtained from a fragmented Felis catus genomic sequence and the National Center for Biotechnology Information’s Cross Species Megablast of multiple species’ FXII gene sequences. Fourteen exons ranging in size from 57 to 222 base pairs were confirmed spanning 8 Kb on chromosome A1. The 1828–base pair feline FXII messenger RNA (mRNA) sequence contains an open reading frame that encodes a protein of 609 amino acids with high homology to human FXII protein. Total RNA and mRNA purified from liver tissue of 4 wild-type/normal and 8 FXII-deficient cats confirmed the predicted mRNA sequence and identified one important single-nucleotide polymorphism (SNP). A single base deletion in exon 11 of the FXII coding gene in our colony of cats results in deficient FXII activity. Translation of the mRNA transcript shows a frame shift at L441 (C441fsX119) resulting in a nonsense mutation and a premature stop codon with a predicted 560–amino acid protein. The mutant FXII protein is truncated in the 3′ proteolytic light chain region of the C-terminus, explaining its loss of enzymatic activity. This study is the first molecular characterization of the feline FXII gene and the first identification of an FXII mutation in the domestic cat, providing insights into the origin and nature of feline FXII deficiency.
Deficiency of factor XII (FXII or Hageman factor) was first described in 1955 in a human patient who lacked plasma coagulant activity. 31 In the intervening time, FXIIs from multiple species have been genetically characterized and various mutations have been reported in humans. Although FXII deficiency does not present as a bleeding tendency, recent evidence indicates FXII may be important in maintaining blood clot stability and pregnancy. 22,25,32 A feline FXII-deficient case was first described in 1977, 10 and later Kier et al 18 established a colony in the early 1980s to study FXII deficiency. Today, these offspring continue to provide information about the in vivo role of FXII and are the source of this study’s molecular characterization of a mutation in the FXII gene with a similar autosomal recessive inheritance pattern and coagulation profile as seen in humans. Studies in human populations indicate that the prevalence of moderate to severe FXII deficiency is between 2% and 5% of a given white population. 11 Similar to humans, the prevalence of FXII deficiency in domestic cats of the United States is 2%. 3
Factor XII is primarily produced in the liver and circulates in the plasma as an inactive precursor enzyme or zymogen. 40 The FXII protein is a 2-chain molecule consisting of a heavy chain (50 kiloDaltons or kD) and a light chain (30 kD) connected by a disulfide bond. 6 When the heavy chain contacts negatively charged surfaces or misfolded proteins, the FXII zymogen changes conformation to expose its active site (on the light chain) in a process termed autoactivation. 34,35 Other coagulation factor zymogens, prekallikrein and factor XI, bind at the exposed serine protease active site and become activated to kallikrein and factor XIa, respectively. Because autoactivation of FXII is the initial step for in vitro coagulation induction, it has been labeled a contact activation factor and initiator of the intrinsic coagulation pathway. 34,35 Activated FXII (FXIIa) has also been shown to bind to cell surfaces via its heavy chain and participate in other biological processes such as inflammation and fibrinolysis, but little is known about the extent of its participation and its importance in pathological outcomes. 14,22,34,37 The heavy chain has amino acid regions that are homologous to fibronectin type I and type II, epidermal growth factor, and kringle structural domains. 27 Fibronectin type I and II domains can bind extracellular matrix proteins and are thought to be involved in the cell-mediated binding of FXIIa. 2,6,36
In humans, congenital FXII deficiency is caused by a variety of genetic defects that result mostly in a loss of functional and identifiable FXII protein. The most common defect is a single 46 “c” to “t” (46C>T) nucleotide substitution in the promoter region of exon 1 on 1 allele (heterozygous) or both alleles (homozygous). 17,39 Whites have a 20% incidence of the nucleotide 46T substitution (2%–6% incidence for a 46T substitution on both alleles), resulting in a decreased translation of FXII due to disruption of the translation initiation site. 17 Individuals affected with this mutation can have FXII activity and antigen levels at or slightly below standard normal ranges, and hence they may not be identified with a FXII deficiency. 8,17,39 Very few mutations result in detectable FXII antigen (referred to as cross-reacting material positive), and these rare mutations tend to occur in exons 9 through 14. 12,17,23,24 Three defects have been reported as single-nucleotide base deletions and subsequent frame shifts from mutations in either exon 12 (c.10590delC and c.10586delG) 33 or exon 14 (splice site mutation 11397G>A). 33 One single-nucleotide base substitution in exon 11 (c.10372 G>A) 33 and 1 point mutation in exon 10 resulted in expression of variant FXII proteins in the patients’ bloodstream and in a cell line transfected with the mutant gene. 33,38
We report the coding sequence of the normal cat FXII gene and a mutation in exon 11 of mutant cats that is associated with a decrease in plasma FXII activity. We also report the normal and mutant cat FXII messenger (mRNA) sequences and their predicted protein sequences. Furthermore, in vitro studies of the FXII protein in cat plasmas reveal functional differences in proteolytic activity of normal and mutant feline FXII.
Materials and Methods
Animals
Domestic shorthair cats ages 1 to 10 years, including 2 female (F) and 2 male (M) wild-type cats, 1 F and 3 M cats severely deficient for FXII, and 1 F and 3 M cats moderately deficient for FXII, were used to sequence DNA or RNA from liver in accordance with the procedures outlined in the Guide for Care and Use of Laboratory Animals and a protocol approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill. Our FXII-deficient cats were derived from a colony established by Kier et al, 18 after approximately 11 years and unknown generations from the colony at Kansas University (a gift from Dr Bresnahan). We received 4 severely FXII-deficient and 2 moderately FXII-deficient cats, of which 1 male and 1 female severely FXII-deficient cat were from the same litter. An outbreeding strategy was used to generate heterozygous (HET)/moderately FXII-deficient offspring in which the gifted homozygous (HZY) males were bred to normal (NL)/wild-type females from outside the colony. The heterozygous female offspring of 1 litter were bred to HET male progeny of a separate litter or those HZY/severely FXII deficient to maintain the colony for 2 to 4 generations. Progeny of our fifth generation were derived by breeding 2 HZY male fourth-generation progeny to female wild-type cats from outside the colony to maintain an inbreeding coefficient of 5%. Pedigree studies to document the segregation of the FXII mutation were not performed.
FXII Activity Assays
A modified 1-stage activated partial thromboplastin time (APTT) was used to measure FXII coagulant activity (FXII:c) while a chromogenic peptide (S-2302) was used to measure amidolytic activity. 4,20 A normal platelet-poor 3.8% citrated plasma pool was created using 4 to 5 normal cats (outside and colony sources). A homozygous cat plasma pool created from 4 living colony cats identified through breeding and FXII:c testing was used as a substrate to dilute plasmas to 1:10 and 1:20 for the FXII coagulant assay. Two of the HZY cats used for the pool were genotyped in this study. In total, 100 μl of prewarmed phospholipid reagent (Dade APTT-FSL; Dade Behring, Inc, Newark, DE) containing kaolin was added to 100 μl of diluted plasma and then warmed to 37°C for 3 minutes before adding 100 μl of prewarmed 25 mM CaCl2. A Stago ST4 coagulation analyzer (Diagnostica Stago, Inc, Parsippany, NJ) was used to monitor clotting time in seconds. A standard curve was generated using serial dilutions (1:10, 1:20, 1:40) of a normal cat plasma pool, which had been given an assigned value of 100% activity. Individual samples (assayed in duplicate) were compared with this curve and the activity expressed as a percent of a normal cat plasma pool. We defined a NL cat as having an averaged FXII activity level >85% of a normal cat plasma pool, a HET cat as having an activity of 10% to 85% a normal cat plasma pool, and a HZY cat as having <10% activity of a normal cat plasma pool.
The amidolytic assay measures the cleavage of pNA from the chromogenic substrate H-D-Pro-Phe-Arg-pNA (S-2302) by kallikrein. FXII activation to FXIIa by ellagic acid and phospholipid catalyzes the activation of prekallikrein to kallikrein, which is necessary to generate an absorbance change in the chromogenic substrate. Since the solid-phase activation of FXII initiates the coagulation cascade in vitro, amidolytic activity reflects FXII activation. Chromogenic substrate S-2302 (Dia Pharma Group, West Chester, OH) H-D-Pro-Phe-Arg-pNA was used to test kallikrein activation in most of our colony animals. Briefly, 25 μl of plasma diluted 1:5 with 0.15M barbital, 0.125M sodium chloride (NaCl), 0.25% bovine serum albumin (pH 7.5) was placed in a 96-well microtiter plate with 25 μl of APTT reagent (Dade Behring, Inc) to achieve a final plasma dilution of 1:10. The plate was incubated at 37°C for 45 minutes, and then 50 μl of 0.0005M S-2302 in 0.05M Tris-HCl, 0.05M imidazole, and 0.15M NaCl (pH 8.2) was added to the activated plasma, with wells monitored every 10 minutes at 405 nm for 1 hour. A standard curve generated from serially diluted pooled normal cat plasma was included on the plate to determine a percentage of normal pool activity for each test sample (assayed in duplicate). The normal cat FXII:c range for our colony is 85% to 110%. Normal human plasma was analyzed as an internal control to assess reagent and machine functions.
Molecular Techniques
Four normal cats from outside the colony and 1 normal cat from our colony were tested for FXII coagulant and enzymatic activity and considered normal for FXII levels. Three normal cats were sequenced for coding sequences in genomic DNA (gDNA) and complementary DNA (cDNA) synthesized from mRNA. One normal cat’s complete cDNA was sequenced from –2 to +1849. One severely FXII-deficient (HZY) cat was sequenced for all gDNA exons and the cDNA of mRNA from –1 to +1838, while 3 other severely deficient cats were sequenced for exons 3 to 14. Four cats moderately deficient for FXII (HET) were sequenced for exons 1 to 4 and 7 to 14 and cDNA of mRNA from +8 to +227 and +1261 to +1830. A summary of the animals used in this study is provided in Supplemental Table S1.
Genomic DNA was extracted from liver samples using the Wizard Genomic DNA Isolation Kit (Promega, Madison, WI) according to the manufacturer’s instructions. The DNA concentration was adjusted to 100 ng/ml for use in polymerase chain reactions (PCRs). The region of the cat genome 29 identified as being a putative ortholog for FXII (ie, F12; cat chromosome A1: AANG02102688.1:5551…13625) was retrieved using GARFIELD (http://lgd.abcc.ncifcrf.gov/cgi-bin/gbrowse/cat/). 30 The cat sequence was aligned to GenBank sequences representing transcripts for this gene from human (NM_000505), mouse (NM_021489), rat (XM_225172, currently NM_001014006), cow (XM_883314, currently NM_001075119), and dog (XM_546206). Candidate primers were selected in immediately flanking intronic sequences located 50 bases upstream/downstream of acceptor/donor splice sites to extend through each identified exon pair (7 exon pairs in total). Primer sequences and PCR conditions for each pair are defined in Table 1. DNA was amplified on a PerkinElmer (Waltham, MA) GeneAmp PCR Systems 9700 thermocycler using the HotStar HiFidelity Taq polymerase kit (Qiagen, Valencia, CA). The assay used a 25-μl reaction mixture containing 1 to 2 μg of genomic DNA, 4 pmol of forward and reverse primers, 5× buffer containing 25 mM MgSO4, 0.75 units per reaction of HotStar HiFidelity Taq polymerase, and PCR water to a final volume of 25 μl. Cycles varied according to each primer set Tm: 95°C for 15 minutes of initial denaturing and enzyme activation, then step-down amplifications of 0.3°C to 1.0°C decreases per amplification step at 10 cycles per temperature were performed (Table 1). For example, the primer set for gDNA exons 7 to 8 had the following: 95°C for 15 minutes; 94°C for 45 seconds, 60.3°C for 45 seconds, 72.0°C for 1 minute × 10 cycles; 94°C for 45 seconds, 60.0°C for 45 seconds, 72.0°C for 1 minute × 10 cycles; 94°C for 45 seconds, 59.7°C for 45 seconds, 72.0°C for 1 minute × 10 cycles; 72.0°C for 10 minutes; and 4°C infinity hold.
Primers Used for Genomic DNA Amplification of Exons and Exon/Intron Boundaries of Feline F12.

PCR, polymerase chain reaction.
Total RNA was extracted from liver tissue samples collected and immediately frozen in liquid nitrogen and then stored at –70°C until further processed with the Absolutely RNA RT-PCR miniprep kit (Stratagene, La Jolla, CA) or the Purelink Total RNA Purification System (Invitrogen/Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions. One NL, 1 HET, and 1 HZY cat total RNA were further purified for mRNA products using the GenElute mRNA Miniprep Kit (Sigma-Aldrich, St Louis, MO). All RNA samples were treated with 1 unit DNase I (Stratagene) per microgram RNA for 15 to 30 minutes at room temperature, then heated to 65°C for 10 minutes prior to reverse transcription. Two-step reverse transcription PCR (RT-PCR) of 1 μg of liver RNA was performed using the ImProm-II Reverse Transcription System (Promega, Madison, WI) in a 20-μl reaction volume containing 5 mM MgCl2, 0.5 mM deoxyribonucleotide triphosphates (dNTPs), 20 units of recombinant RNasin Ribonuclease Inhibitor, 1 μl of reverse transcriptase, 0.5 μg of random hexamers or oligo (dT) primers, and PCR water to a final volume of 20 μl. PCR was performed in a 25-μl reaction volume containing 5× reaction buffer, 0.3 mM dNTPs, 4 pmol of forward and reverse primers, 5× Q-Solution (Qiagen), 1.5 units per reaction of Taq polymerase, 3 to 4 μl of cDNA, and PCR water to a final volume of 25 μl. A touch-down procedure was used as follows: 95°C for 15 minutes (HotStar HiFidelity Taq polymerase; Qiagen) or 2 minutes (GoTaq Hot Start polymerase; Qiagen); 94°C for 30 seconds, touch-down 62.5°C for 30 seconds, then decrease 0.5°C per cycle × 5 cycles, 72°C for 30 seconds; and then 30 cycles of 94°C for 15 seconds, 60°C for 45 seconds, and 72°C for 45 seconds, with final holds of 72°C for 7 minutes and then 4°C infinity hold (see Suppl. Table S2 for each primer set’s conditions). PCR products were visualized by electrophoresis on 1.5% low electroendosmosis agarose gel. Bands were excised and cleaned using Qiagen’s QIAEXII kit. Forward and reverse sequence data for each PCR amplicon were generated at the UNC-CH Automated DNA Sequencing Facility and the University of Michigan DNA Sequencing Core. Sequences were analyzed using Sequencher 4.8 software (Gene Codes, Ann Arbor, MI). The prediction program Clustal omega version 1.2.0 was used to align cDNA and protein sequences.
Protein Isolation and Identification Techniques
Plasma mixing studies and inhibition of feline FXII
To define the functional enzymatic activity of normal and mutant feline FXII, we performed plasma mixing studies using human prekallikrein-deficient (PKd) plasma (Sigma Diagnostics, St Louis, MO) and FXII-deficient plasma (George King Biomedicals, Overland Park, KS), corn trypsin inhibitor (CTI; Enzyme Research Laboratories, South Bend, IN), and a monoclonal mouse anti–human FXII heavy chain antibody, B7C9, directed to amino acids p.I19–V38 and p.T153–R172 of the human FXII protein 5,28 (a gift from Dr Robin Pixley) with cat plasmas prior to assaying coagulant activity using the Stago ST4 and the modified APTT procedure. Human PKd plasma was used as a diluent instead of a HZY cat plasma pool to assess for a prekallikrein deficiency in our colony. Antibodies or CTI were added to determine their ability to specifically inhibit feline FXII coagulant activity. Plasma duplicates were incubated 15 to 60 minutes at 37°C with each inhibitor prior to performing the APTT. One normal cat, 3 HZY cats, and the HZY cat plasma pool were evaluated in the prekallikrein studies. The HZY cat plasmas were diluted 1:2 with human PKd plasma, while normal cat plasma required 1:10 to 1:40 dilutions because the extremely short clotting time observed with normal cat plasma (<18 seconds at a 1:2 dilution) would not be detected on the ST4. Prekallikrein sufficient or deficient was determined as the ability to correct the prolonged APTT clotting time in less than 60 seconds (Table 2). Inhibition of FXII activity by CTI or B7C9 mouse monoclonal antibody was determined as the ability to prolong the APTT by double the control value.
Mixing and Inhibition Studies of Cat Colony Plasmas.
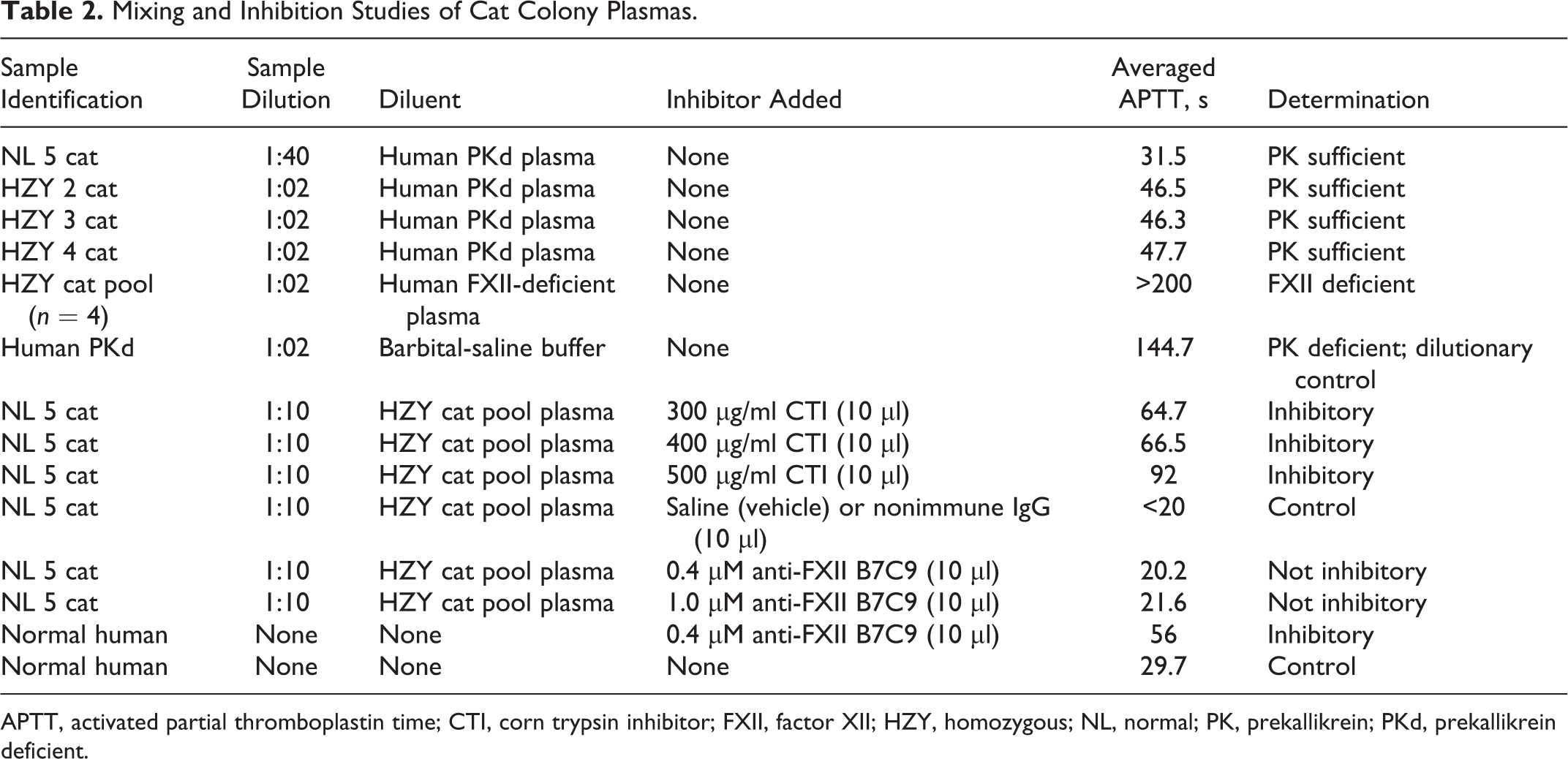
APTT, activated partial thromboplastin time; CTI, corn trypsin inhibitor; FXII, factor XII; HZY, homozygous; NL, normal; PK, prekallikrein; PKd, prekallikrein deficient.
Protein electrophoresis and Western blotting
Diluted plasma samples from a normal cat and homozygous FXII-deficient cat were subjected to reducing conditions and 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) prior to electroblotting onto nitrocellulose or polyvinylidene fluoride. The blotted antigens were detected with 2 polyclonal (pAb) anti–human FXII antibodies (purchased from Cedarlane, Burlington, Ontario, Canada [CL20055A] and Affinity Biologicals, Ancaster, Ontario, Canada) and a monoclonal “B7C9” heavy chain–specific anti–human FXII antibody (a gift from Dr Robin Pixley) as described by Pixley and Colman. 27 Prestained (P8748 [Sigma]; Kaleidoscope [Bio-Rad, Hercules, CA]) molecular weight (MW) standards were used to determine the size and migration of detected antigens. Secondary antibodies of either rabbit anti–goat IgG or rabbit anti–mouse IgG conjugated to peroxidase (Sigma-Aldrich) or rabbit anti–mouse alkaline phosphatase (Jackson Laboratories, Bar Harbor, ME) were detected with Vector Laboratories (Burlingame, CA) NovaRed or Sigma BCIP/NBT substrates. Additional studies of feline plasmas involved 2-dimensional difference in gel electrophoresis (2-D DIGE) with subsequent Western blot (WB) analysis. 41 Normal feline plasma stained with CyDye3 (green) and HZY feline plasma stained with CyDye 5 (red) were mixed together at equal ratios and then run on a pH 3 to 10 nonlinear IEF followed by 10.5% SDS-PAGE for the second dimension prior to imaging on ImageQuant software (GE Healthcare, Piscataway, NJ). Electroblotting and immunodetection were performed as described, using a peroxidase-conjugated rabbit anti–goat secondary antibody (A5420; Sigma-Aldrich) and enhanced chemiluminescence (ECL) detection kit (Amersham ECL; GE Healthcare).
Protein isolation methods
Normal feline FXII protein was isolated on a pAb (Cedarlane) goat anti–human FXII-CNBr Sepharose immunoaffinity column prepared as described by Kier et al. 19 Briefly, 20 to 50 ml of fresh-frozen citrated plasma (pH 6.0) was thawed at 37°C and treated with 100 μg/ml soybean trypsin inhibitor (SBTI) and 50 μg/ml polybrene (hexadimethrine bromide; Sigma-Aldrich). To remove contaminating proteins and isolate FXIIa, plasmas were applied to benzamidine-HCl agarose columns and fractions were collected. Fractions with detectable FXII activity (using the APTT assay with HZY cat pool plasma) were dialyzed overnight at 4°C in loading buffer (0.01M Tris, 0.5M NaCl, 100 μg/ml SBTI, and 50 μg/ml polybrene [pH 7.5]) and further purified using the goat anti–human FXII-CNBr immunoaffinity column. Bound feline FXII was eluted with 0.05M glycine/50% ethylene glycol buffer (pH 3.2) on the immunoaffinity column with a linear gradient of 0.15 to 1.5M sodium chloride. Then, 2- to 5-ml fractions were collected with an LKB optical fractionator, dialyzed/desalted, concentrated, and tested for FXII antigen (WB) and activity (APTT). The anti–FXII-CNBr immunoaffinity column was stripped twice with 10 ml of 2M guanidine hydrochloride and re-equilibrated with 0.01M Tris, 0.5M NaCl, 100 μg/ml SBTI, and 50 μg/ml polybrene (pH 7.5) buffer prior to use. All fractions of normal or HZY cat plasma with 280-nm absorbance readings above 0.1 were tested for antigen and activity. The column fractions were reduced, loaded at 5 μg total protein, electrophoresed, and immunoblotted with anti–human FXII antibodies.
Results
The feline FXII gene consists of 14 exons and 13 introns, which results in a predicted protein of 609 amino acids (AA) having high sequence similarity to human (75% exons, 75% amino acid) and canine (85% exons, 85% amino acid) FXII, as shown in Supplemental Table S3. The coding sequence for 1 normal cat FXII (accession number GQ_981174) and 1 mutant cat FXII (accession number GQ_981175) was determined and submitted to GenBank. No polymorphisms were found in the exonic regions using the cat Contig6175a.247 sequence as an alignment reference for our normal cat samples. A mutation consisting of a deletion of 1 “c” nucleotide in exon 11 (c.1321delC) was found in all FXII-deficient cats tested (Suppl. Table S4 box and Suppl. Fig. S1 arrow for identified base). As expected, the severely FXII-deficient cats were homozygous, while moderately affected cats were heterozygous for the mutation.
Analysis of the sequenced feline RNAs (cDNA) indicates the FXII gene encodes for a mature protein of 609 AA and is predicted to contain all the characteristic structural domains reported in human FXII—that is, a type I and II fibronectin domain, 2 epidermal growth factor–like domains, a kringle domain, a proline-rich domain, and the typical trypsin-like serine protease domain of the light chain. The light chain’s catalytic triad of H404, D453, and S557 predicted for feline (Suppl. Table S3, boxed AA) has all 3 amino acids aligning with the human translated sequence (H412, D461, and S562 in humans) in addition to the proteolytic cleavage site of the light chain at R364–V365 in cat (Suppl. Table S3, underlined residue) or R372–V373 in human. However, only 1 of the 2 predicted kallikrein cleavage sites in human (R353–N354 and R362–L363) FXII 12,27 shares homology with the cat sequence, as illustrated in Supplemental Table S3 (bolded and italicized AA). Using the prediction program SignalP3.0, a signal peptidase cleavage site was identified between amino acid position S16 and A17 in the feline FXII protein, indicating the secreted protein would be 593 amino acids in length. 1 The deletion mutation, c.1321delC, causes a frame shift and missense mutation at L441C (C441fsX119) with a premature stop codon at position 560 in the protein. Exons 11 through 14 encode most of the trypsin-like serine protease domain (active site). The genomic single base deletion would likely result in premature truncation of feline FXII protein.
Analysis of the predicted feline FXII proteins revealed many interesting characteristics. The theoretical isoelectric point (pI) and MW for normal feline FXII are 6.02 and 67.4 kD, respectively. The predicted/theoretical pI and MW for the mutant protein are 9.01 and 62.4 kD, respectively. 9 Predictions of arginine cleavage sites (autoactivation or light chain proteolytic cleavage sites) in human FXII (Suppl. Table S3, bold and italicized AA) suggest that normal and mutant feline FXII have 1 site at p.R354 and 1 site at p.R372–V373 (site sequence RLSSLSR|VV) similar to human (Suppl. Table S3, underlined AA). 7,27
Protein studies of plasma and purified feline FXII confirm some of the predicted characteristics. According to WB analyses with various antibodies directed to human FXII, the normal feline protein migrates at about 66 kD (data not shown). Table 2 illustrates that normal feline FXII has prekallikrein activity, and its coagulant activity can be inhibited with CTI. Our results suggest the monoclonal heavy chain anti–human FXII antibody (directed against amino acids p. I19–V38 and p.T153 through R172) does not have inhibitory activity at the concentrations evaluated. Human studies show enzymatic inhibition properties using both agents. 16,28 When the purified and concentrated fractions of FXII protein from normal cats were added to a homozygous cat plasma pool, the APTT was corrected to near-normal values (from >200 seconds corrected to 33.6–43.1 seconds), indicating that the purified FXII was intact and active (data not shown). Testing of the mutant factor XII protein isolated from deficient cat plasma revealed no correction of the APTT (>200 after adding purified mutant FXII), suggesting this abnormal protein lacks FXII catalytic activity reflective of a primary defect in FXII protein (Suppl. Table S3). Factor XII protein was detected in mutant cat plasma samples (Fig. 1a, red) and purified fractions using the human-directed antibodies (data not shown). The apparent size of the zymogen mutant FXII protein is approximately 66 kD, with its heavy chain of appropriate size (∼50 kD) recognized by the monoclonal heavy chain antibody B7C9. Under isoelectric focusing and reduced SDS-PAGE (2-D DIGE) conditions, it appears the mutant FXII protein expressed in the plasma has a slightly different migration pattern (Fig. 1a, circled, red spot). Western blot analysis with a polyclonal anti–human FXII antibody suggests the 2 identified spots (circled) in Fig. 1b should be FXII proteins corresponding to the identified gel proteins (Fig. 1a, circled red and green spots).

Two-dimensional difference in gel electrophoresis (2-D DIGE) of CyDye-labeled citrated plasmas. One normal cat (green, “n”) and 1 homozygote cat (red, “m”) plasma sample were mixed and then separated by 2-D DIGE and visualized in Fig. 1a. The circled spots correspond to suspected factor XII proteins detected by Western blot techniques shown in Fig. 1b. An enlarged view of the area of interest is shown in the upper left panel.
Discussion
This study has successfully identified a genomic mutation that would likely result in premature truncation of feline FXII. The corresponding mutation is present in abnormal feline mRNA transcripts coding for FXII. The mutation identified in exon 11 of our FXII-deficient cat colony is the first molecular characterization of this defect in domestic shorthaired cats. Although no identical nonsense mutation has been described in humans, other single base deletions in exons 10, 12, and 14 result in similar phenotypes. 33,38 Studies of our HZY cat plasmas indicate that a catalytically inactive cross-reacting material-positive protein is expressed, as detected by various commercially available polyclonal and monoclonal anti–human FXII antibodies. The active site and heavy chain binding site appear to be similar in feline and human FXII according to their amino acid sequences and protein studies. In humans, the heavy chain has high homology with urokinase and tissue plasminogen activator, 6 along with type II fibronectin regions, which may contribute to FXII’s binding to cellular surfaces and collagen. 2,36 We evaluated whether prekallikrein activation to kallikrein was altered in our cat colony via plasma mixing studies. By using prekallikrein-deficient human plasma mixed with mutant cat plasma (suspected to be FXII deficient), we were able to test whether cat kallikrein could activate human high molecular weight kininogen (HK) and ultimately human FXI. Normal cat plasma has high prekallikrein-kallikrein activity, illustrated in Table 2, in that the sample (NL 5 cat) required a high dilution (1:40) by human prekallikrein-deficient plasma to achieve a measurable clotting time of 31.5 seconds. The HZY mutant FXII cat plasmas had less kallikrein activity since their plasma samples required a dilution of 1:2 or 50% to achieve a measurable clot. Factor XII activation by ellagic acid in normal cat plasma generates high amounts of activated kallikrein, which appears to cross-activate human HK and FXI present in the mixture. However, the mutant cat FXII cannot generate high levels of kallikrein and perhaps requires human FXII activation to cross-activate the cat prekallikrein and/or directly catalyze human FXI. It is possible the human FXIIa protein (present in the human PKd plasma) may have a reduced binding capacity for cat prekallikrein that results in an impaired kallikrein generation. When mutant cat plasma (HZY pooled plasma, Table 2) was mixed with human FXII-deficient plasma, no clotting activity occurred. These results reconfirm the mutant cat colony defect is in the activation of FXII.
Our studies suggest the expressed mutant cat FXII protein retains a portion of its C-terminal light chain region yet lacks the proper active site residues D453 and S557, which agrees with the predicted protein characteristics. However, we cannot eliminate the possibility that our antibodies are detecting some other cross-reactive protein capable of activating normal FXII when tested in an APTT assay, as shown in Table 2. Certain FXII-deficient humans have mutations that result in amino acid alterations occurring in the C-terminal region of the protein and can result in a loss of detectable protein or an expressed protein lacking enzymatic function. A human FXII variant has a G>A nucleotide change in the 3′ splice acceptor site of exon 14 (c.11397 G>A) that causes the exon 13 to exon 14 splicing product to lack 1 nucleotide. This splice site mutation is believed to result in an unstable truncated protein that is cross-reactive negative. 33 Factor XII Washington DC variant (p.C571S) loses the disulfide linkage between C571 and C540, which maintains a loop structure containing the active site serine (S544). The loss of this looping alters the conformation of the active site serine or the secondary substrate-binding site, thereby causing a loss of enzymatic activity. 24 Other protein-producing mutations in FXII-deficient patients have also been identified. An R353P amino acid substitution in exon 10 results in a loss of the kallikrein cleavage site seen in a FXII Locarno variant, 12 and a substitution (W486C) in exon 12 results in a reduced translation and a reduced secretion of FXII due to incorrect protein folding seen in FXII Mie-1. 42 All of these cross-reacting material-positive variants are rare occurrences found in the FXII-deficient human population. Two frame shift–causing mutations found in exon 12 (c.10568delG and c.10590delC) in humans result in less than 1% detectable FXII protein, which is assumed to be unstable. 33 The recently identified FXII Ofunato variant results in a K346N substitution in exon 10 and 5% detectable FXII protein level due to incomplete degradation via the endoplasmic reticulum quality control machinery. 38 Most likely, the severity of the predicted changes to our colony’s mutant FXII protein would result in an unstable misfolded protein that is subject to proteasome degradation. 21,38 Further studies of the mutant cat FXII protein are needed to determine the expression status and sequence of the predicted protein.
In humans, recurrent pregnancy loss in the first trimester has been associated with FXII deficiency. 25 Other studies have found anti-FXII antibodies may alter platelet-FXII binding and promote pregnancy loss in humans. 13,15 Interestingly, Inomo et al 13 found 76.5% of their recurrent pregnancy loss subjects had antibodies that recognized amino acids 1 to 30 in the heavy chain of FXII (a region where monoclonal antibody B7C9 binds).
Although associations have been found with FXII deficiency and recurrent pregnancy loss in humans, the transgenic mice models of FXII deficiency have reported no adverse pregnancy outcomes. 26,32 We evaluated the percentage of kittens born dead (stillborn) per litter in our colony and found HZY first-time mothers had significantly more stillborn kittens (67%) compared with HET (23%) and NL (17%) first-time mothers (P < .05; 1-way analysis of variance). We caution that inbreeding and environmental factors can affect pregnancy outcomes, 18 and we have no cause of death or further evidence that FXII was related to the high kitten stillborn rate in HZY mothers.
In conclusion, new insights into the molecular characterization of feline FXII have been gained. Identification of the FXII genetic defect causing a loss of coagulant and amidolytic activity in a research colony of cats will aid in future analyses investigating the in vivo role of FXII.
Footnotes
Acknowledgement
We thank Ms Pamela McElveen, Dr Radwan Abu-Issa, Dr Dougald Monroe, and Dr David Klapper for their assistance on this project.
Authors’ Note
NCI-Frederick is accredited by AAALAC International and follows the Public Health Service Policy for the Care and Use of Laboratory Animals. Animal care was provided in accordance with the procedures outlined in the Guide for Care and Use of Laboratory Animals (National Research Council; Washington, DC: National Academies Press; 1996) and in accordance with a protocol approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government. Funded by NCI Contract N01-CO-12400.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.