
Abstract
Mucopolysaccharidosis (MPS) type IIIB was diagnosed in 14 juvenile emus (Dromaius novaehollandiae), ages 3 weeks to 6 months, based on pathological and biochemical analyses. The animals had a history of neurological signs or sudden death; one of the birds with neurological signs and 3 others experienced acute hemoabdomen. Histopathologically, neuronal swelling and vacuolation in the cerebrum, cerebellum, brainstem, and spinal cord (80%–92%); retina (100%); autonomic ganglia of the intestine (71%); gizzard (50%); adrenal gland (27%); and ear (50%) were noted in affected but not healthy emus. Cytoplasmic vacuoles were also observed in the pancreas, liver, intestine, adrenal glands, and kidneys. The intracytoplasmic inclusions were periodic acid–Schiff and Luxol Fast Blue positive, consistent with a storage disease. Foamy macrophages infiltrated the liver, intestine, tunica media of the aorta, and spleen. By transmission electron microscopy, typical lamellated cytoplasmic bodies were detected in neurons of the brain and retina, while electron-dense bodies consistent with glycosaminoglycan inclusions were observed in hepatocytes and/or hepatic macrophages. The livers of the 2 affected emus studied contained large amounts of heparan sulfate, which is suggestive of MPS type III. Compared with normal controls, hepatic and serum α-N-acetylglucosaminidase activity was very low (<8% of control), while other enzyme activities were normal to increased in the 2 affected emus studied. Moreover, affected emus were homozygous for a 2-bp deletion in the NAGLU gene. This study characterizes the pathology of MPS type IIIB in emus, which is one of the rare inborn errors in birds, showing the homology of this condition to Sanfilippo syndrome in humans.
Keywords
Lysosomal storage diseases (LSDs) are a group of inborn errors of metabolism resulting from deficiencies in specific lysosomal acid hydrolases. 22 LSDs are usually chronic and progressive with a wide spectrum of clinical severity depending on whether the central nervous system (CNS), musculoskeletal system, and other organ systems are affected. Cells most active in turning over the substrate or long-lived postmitotic cells (eg, neurons, cardiac myocytes) are most vulnerable to storage. The fixed and mobile macrophages are also common target cells. 11,37
Numerous LSDs have been described in humans, 37 and homologous diseases have been found in various domestic species and zoo animals. 21 Among avian species, glycogen storage disease II (Pompe disease) in Japanese quail 36 and GM2 gangliosidosis in American flamingo 32,57 have been described. Interestingly, mucopolysaccharidosis (MPS) type IIIB in emus 1,18,45 has been characterized at the molecular level, while a gangliosidosis 4,5,17 and an undetermined LSD 31 in emus have also been suspected based on pathology.
Mucopolysaccharidoses involve defects in the breakdown of glycosaminoglycans (GAGs) 39 such as dermatan sulfate (DS), heparan sulfate (HS), keratan sulfate (KS), and chondroitin sulfate (CS), which are extremely large and complex extracellular matrix molecules. Eleven enzymes are involved in their degradation, 39 and 7 types of MPS (MPS I, II, IIIA, IIIB, IIID, VI, and VII) have been reported in domestic animals. 20,21,26
Mucopolysaccharidosis type III, also known as Sanfilippo syndrome, results from the absence of 1 of 4 lysosomal enzymes involved in the degradation of heparan sulfate: heparan sulfate sulfamidase (SGSH; MPS type IIIA), α-N-acetylglucosaminidase (NAGLU; MPS type IIIB), acetyl-CoA:α-glucosaminide-N-acetyltransferase (GNAT; MPS type IIIC), and N-acetylglucosamine-6-sulfatase (GNS; MPS type IIID). 39,47 In the absence of any of these enzymes, undegraded or partially degraded heparan sulfate accumulates in lysosomes and is excreted in urine. 21 MPS type III causes progressive neurological signs in association with skeletal anomalies and hepatosplenomegaly. 37,56 In humans, the natural course of the neurological disease is similar in all subtypes: initially aggressiveness and hyperactivity, later progressing to mental retardation and, ultimately, CNS degeneration, 47 although an attenuated course with late presentation may also occur. 38 In addition to humans, naturally occurring MPS type IIIA has been described in Dachshund 16,25 and New Zealand Huntaway dogs, 24,56 type IIIB in Schipperke dogs 13 –15 and cattle, 30 and type IIID in goats. 27,28,46 Here, we describe the clinical, pathological, biochemical, and molecular features of emus affected with MPS type IIIB.
Materials and Methods
Birds: History and Clinical Presentation
Fourteen young emus (age range: 3 weeks to 6 months; 4 males, 6 females, others unknown) with a history of neurological signs or sudden death were submitted to the California Animal Health and Food Safety Laboratory System (Fresno Branch) of the University of California, Davis for diagnosis. The birds originated from farms in California (8), Oklahoma (4), Arizona (1), and Texas (1). Four birds in this study were offspring from 1 particular breeding pair in California that had additional 4 emus with unexplained juvenile deaths. The birds from Oklahoma were offspring from 2 breeding pairs of the same breeder source, with 2 birds per pair. Archived serum and tissue samples as specified below from healthy emus served as controls. The studies were approved by the Institutional Animal Care and Use Committees of the California Animal Health and Food Safety Laboratory System of the University of California, Davis and the School of Veterinary Medicine, University of Pennsylvania.
Necropsy, Blood and Tissue Samples, Histopathology, and Transmission Electron Microscopy
Complete necropsy and histopathologic examinations were performed on all birds. Tissue samples were fixed in 10% neutral buffered formalin and routinely processed (paraffin embedded, sectioned at 5 μm thickness, and stained with hematoxylin and eosin [HE]) for histopathological examination. The histochemical stains periodic acid–Schiff (PAS) and Luxol Fast Blue (LFB) were performed on representative sections. Serum and liver samples were collected freshly and frozen from 2 birds for biochemical and molecular studies. No blood tests were performed, but blood smears were made from 2 birds.
For transmission electron microscopy (TEM), small pieces (1 mm3) of brain, retina, and liver from 2 emus, both offspring of the same breeding pair, were fixed in 2.5% glutaraldehyde, postfixed in osmium tetroxide, dehydrated, and embedded in Epon. Ultrathin sections (70 nm) were stained with uranyl acetate and lead citrate.
Biochemical and Molecular Genetic Analyses
Sample processing
Freshly frozen liver samples from 3- and 20-week-old affected emus and two 12-week-old healthy unrelated emus were studied by the Metabolic Genetics Screening Laboratory of Section of Medical Genetics, School of Veterinary Medicine, University of Pennsylvania (PennGen). In addition, serum samples were obtained from 1 breeding pair (male and female), from which 4 affected emus in the study originated, and from 6 healthy emus. All specimens were frozen prior to analysis.
Aliquots of hepatic tissue (2 × 2 × 3 mm) were homogenized in 1 ml of 0.9% saline containing 0.2% Triton X-100 and sonicated for 20 seconds in an ice bath. The samples were then frozen at –80°C overnight and quickly thawed in a 37°C water bath, and the cellular debris was pelleted by centrifugation at 10 000 g for 10 minutes. Supernatants were then analyzed for GAGs, lysosomal enzymes, and total protein.
GAG analysis
The MPS spot test, 6 typically used with urine samples as a screening test for MPS, was applied on hepatic supernatants, and quantitative GAG analysis was performed according to the manufacturer’s protocol (Biocolor Ltd, Carrickfergus, UK). The GAGs were further separated by cellulose acetate membrane electrophoresis 55 with a standard containing CS, DS, and HS.
Lysosomal enzyme assays
Three panels of lysosomal enzyme activities, totaling 15 enzymes, were assayed in hepatic supernatants for the specific diagnoses of mucopolysaccharidoses, sphingolipidoses, and glycoproteinoses. All enzyme activities were determined by fluorescence assay following standard procedures as previously described for humans, cats, and dogs. 23 Liver enzyme activities were expressed as nmol of 4-methylumbelliferone (4-MU) released per hour per milligram of protein and as a percentage of normal control activity. Supernatant protein concentrations were determined using a kit (Bio-Rad Laboratories, Hercules, CA) and expressed as mg/ml. Serum NAGLU was calculated as nmol of 4-MU per hour per milliliter of serum and also expressed as a percentage of normal control activity.
Genotyping for NAGLU gene mutation
Genomic DNA was extracted from blood and liver tissue. The DNA surrounding the NAGLU mutation in exon 6 was amplified by polymerase chain reaction (PCR), sequenced, and examined for the previously determined NAGLU mutation in emus with MPS type IIIB. 1
Additional diagnostic tests
Serum from 8 animals was tested for avian influenza, avian paramyxovirus 1, and Western and Eastern equine encephalomyelitis. Intestinal content was examined for viral infections 12 in 5 emus. Heavy metals and selenium were determined in liver samples from 7 birds. Routine bacteriology included aerobic culture and enrichment for salmonella in 11 samples and for mycoplasma in 3 samples. These additional test results were negative in all animals.
Results
Clinical Signs
The affected emus were reportedly normal during the first postnatal weeks. Sudden death without previous clinical signs occurred in 5 birds at age 3 to 24 weeks. Other early clinical signs observed were weakness (1/14), anorexia (2/14), and lethargy (2/14). Neurological signs were recognized in 5 of 14 birds and included ataxia, circling, pecking at the air, droopy head, and limping (4/5), abnormal behavior (1/5), curled toes (1/5), walking backward (1/5), torticollis (1/5), and crooked legs (1/5). Clinical information on other birds was lacking.
Pathological Findings
Gross findings
Gross lesions were highly variable but involved different sites of hemorrhage in 12 of 14 emus, including liver rupture with hemoabdomen (4/12) and hemorrhage in the liver (2/12), coelom (2/12), sternum (1/12), neck (1/12), thigh (1/12), and right atrioventricular valve (1/12). A pale carcass (2/14) and fatty liver (2/14) were also noted.
Histopathologic and histochemical findings
Evidence of storage disease was found in the CNS, liver, heart, aorta, intestine, spleen, and eye. The distribution, frequency, and types of microscopic lesions for the 14 affected birds are listed in Supplemental Table S1.
Storage was observed in the cerebrum (12/14 birds), cerebellum (11/12), and brainstem and spinal cord (4/5) (Fig. 1). Neurons were swollen with a pale acidophilic cytoplasm containing fine clear round homogeneous vacuoles. Nissl substance was dispersed by the vacuoles. Within the cerebrum (5/6) and cerebellum (3/6) of 6 birds, the neurons were moderately to severely enlarged due to an increased amount of faintly staining smooth eosinophilic cytoplasm. The cerebellum and nuclei of the medulla oblongata were most commonly affected, while Purkinje cells were spared. In 1 animal, glial cells were also effaced by cytoplasmic swelling and vacuolation. The intracytoplasmic vacuoles were slightly to moderately PAS positive and strongly LFB positive (Fig. 2). In both of the 2 birds with available blood smears, a few monocytes had fine vacuoles in their cytoplasm. No lesions were observed in the spinal root ganglia and peripheral nerves.

In the liver, moderate to severe, multifocal infiltration of large foamy macrophages containing high numbers of fine clear cytoplasmic vacuoles (Fig. 3) (13/14), diffuse lipidosis (9/14), and mild chronic lymphoplasmacytic and histiocytic hepatitis (5/14) was observed. Some vacuoles were clear and contained no stained material, whereas others contained slightly PAS-positive granular material. Four animals had acute (2/4) or chronic (3/4) hepatic hemorrhage, the latter characterized by focal encapsulated well-demarcated hematomas.
Swelling and vacuolation of the autonomic ganglia embedded in the epicardium and epicardial fat (5/13) and macrophages infiltrating the tunica media of aorta (9/10) were observed (Fig. 4). All birds had mild to severe, multifocal to diffuse infiltration of vacuolated macrophages in the mucosa and submucosa of the intestinal tract (Fig. 5), which was associated with swelling and vacuolation of the myenteric/submucosal/subserosal plexuses neurons (10/14) and mild multifocal vacuolation of the smooth muscle cells (8/14).
Moderate to severe, multifocal to coalescing infiltration of vacuolated macrophages in the spleen (7/11), which was rarely associated with hyperplasia of reticular cells (3/11) and amyloid deposition (1/11), was also noted. Neuronal vacuolation was observed in the ganglion cells of the retina (13/13) (Fig. 6), autonomic ganglia of the lungs (1/10), kidney (1/14), and gizzard (5/10) near the adrenal glands (3/11); and in the vestibulocochlear ganglia of the ear (1/2). Pancreatic lesions with specific vacuolation of cells of the islets (4/9), acini (2/9), and ductal cells (1/9) were detected. A diffuse macrovesicular vacuolation of the cytoplasm of adrenal cortical cells was observed (10/11). Fine to large clear intracytoplasmic vacuoles were seen in the epithelial cells of the proximal convoluted tubules of the kidney (12/14) and in the epithelial cell layer of the ciliary body in 1 bird. Sporadic infiltration of vacuolated macrophages was observed in the interstitium of the thyroid (1/8).
Transmission electron microscopic findings
Transmission electron microscopy of the brain and retina revealed numerous membrane-bound lamellated membranous cytoplasmic bodies, consistent with Zebra bodies, in neurons and ganglion cells (Fig. 7). A second type of cytoplasmic inclusion bodies was occasionally detected within retinal cells, consisting of a central granular, highly electron-dense core surrounded by membranous whorls (Fig. 8). Macrophages in the liver contained multifocal well-circumscribed electron-dense homogeneous circular bodies surrounded by stacks of rough endoplasmic reticulum (Fig. 9).
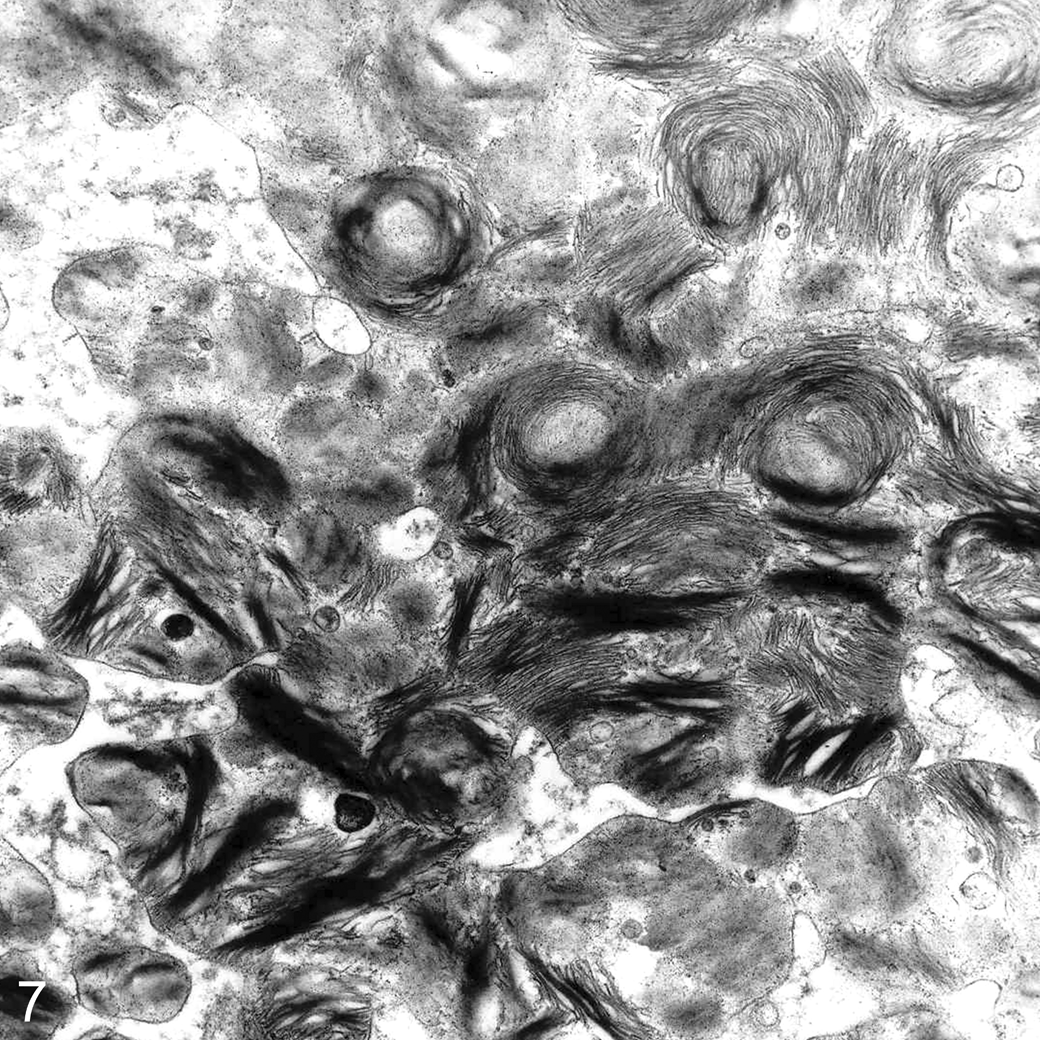
Brain; emu No. 4. Numerous single membrane-bound lamellated membranous cytoplasmic bodies, consistent with Zebra bodies, in a neuron. Transmission electron microscopy. Uranyl acetate and lead citrate.

Retina; emu No. 4. Cytoplasmic inclusion consisting of a central granular highly electron-dense core surrounded by membranous whorls. Transmission electron microscopy. Uranyl acetate and lead citrate.
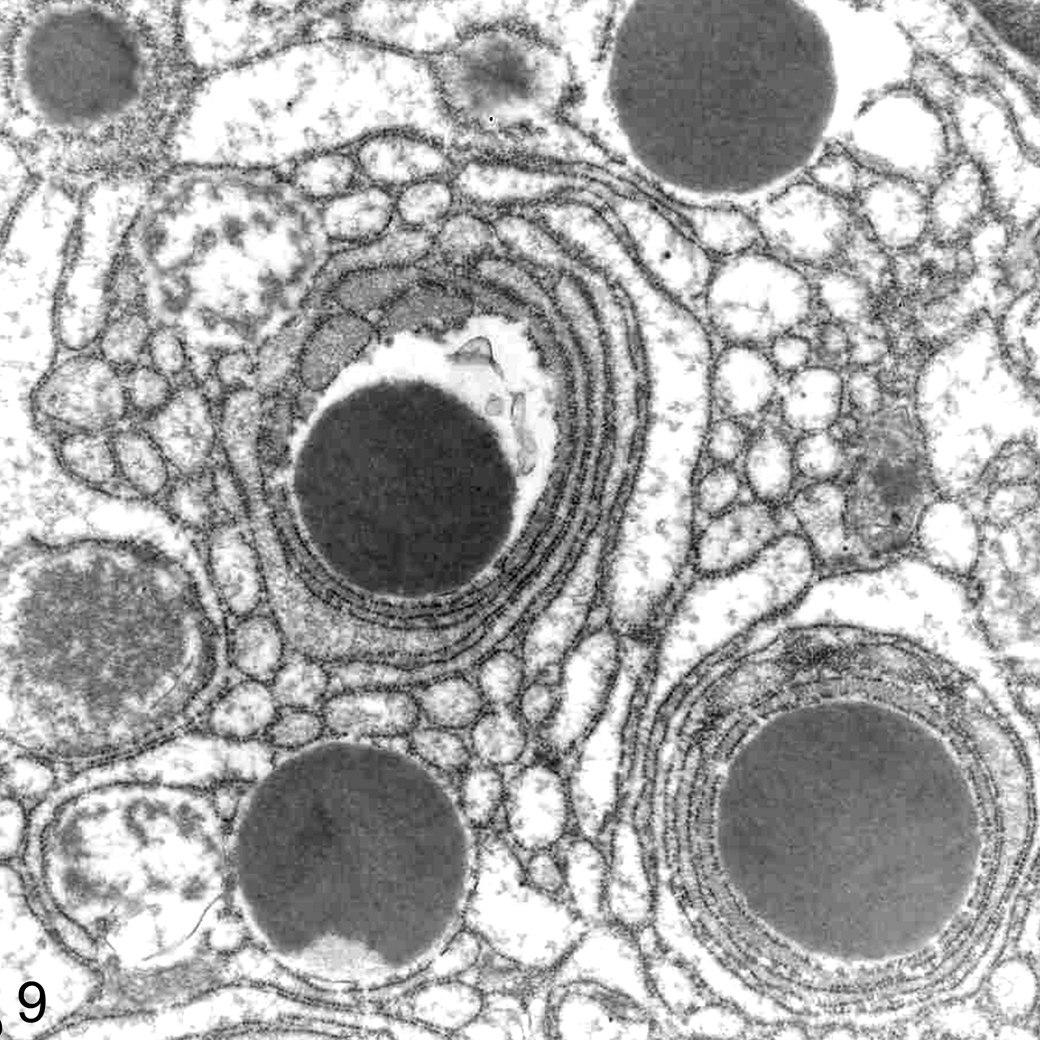
Liver; emu No. 1. Multifocal well-circumscribed but non–membrane-bound electron-dense bodies surrounded by rough endoplasmic reticulum (RER) in the cytoplasm of a foamy macrophage. Transmission electron microscopy. Uranyl acetate and lead citrate.
Biochemical Analyses
Liver glycosaminoglycans (GAGs) analysis
The MPS spot test of the 2 affected emus was strongly positive (Fig. 10A), while that of healthy unrelated emus was completely negative. The quantitation of GAGs revealed 11- to 13-fold increases in supernatants from both affected livers compared with livers from healthy unrelated emus (Fig. 10B). Cellulose acetate electrophoresis showed that the GAGs in hepatic supernatant from the affected emus were exclusively due to HS accumulation (Fig. 10C).
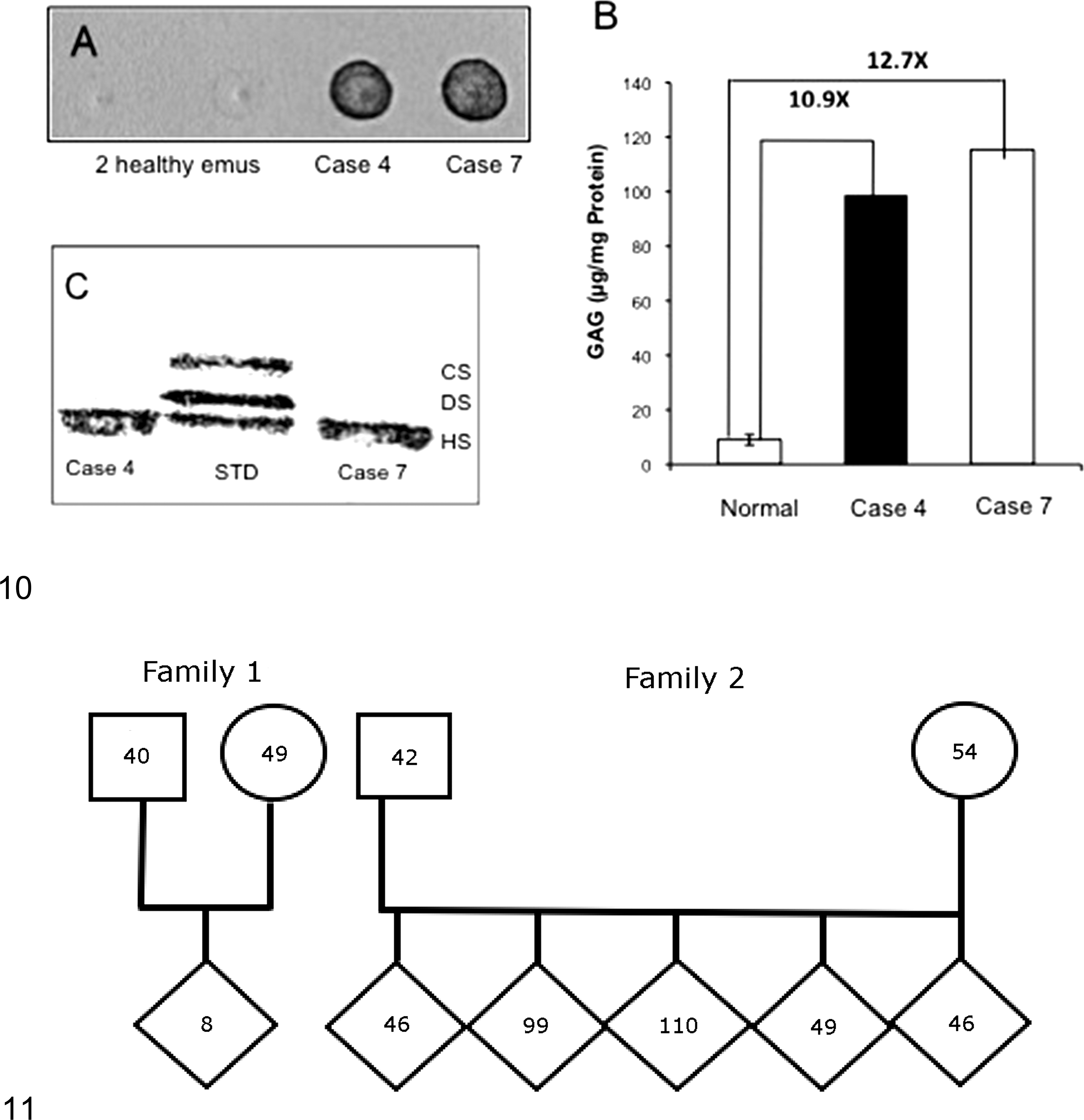
Liver lysosomal enzyme activities and serum NAGLU activities
The results of 15 lysosomal enzyme activities in hepatic supernatants from 2 affected and 2 normal age-matched controls are listed in Table 1. The NAGLU activities of 2 affected emus were 1.4% and 2.9% of normal, respectively. The other enzyme activities involving the GAG degradation pathways were normal or elevated. Moreover, the activities of other lysosomal enzymes related to sphingolipidoses and glycoprotein storage disorders remained unchanged compared with controls.
Hepatic Lysosomal Enzyme Activities in 2 Emus With Mucopolysaccharidosis (MPS) Type IIIB and in 2 Healthy Emus.
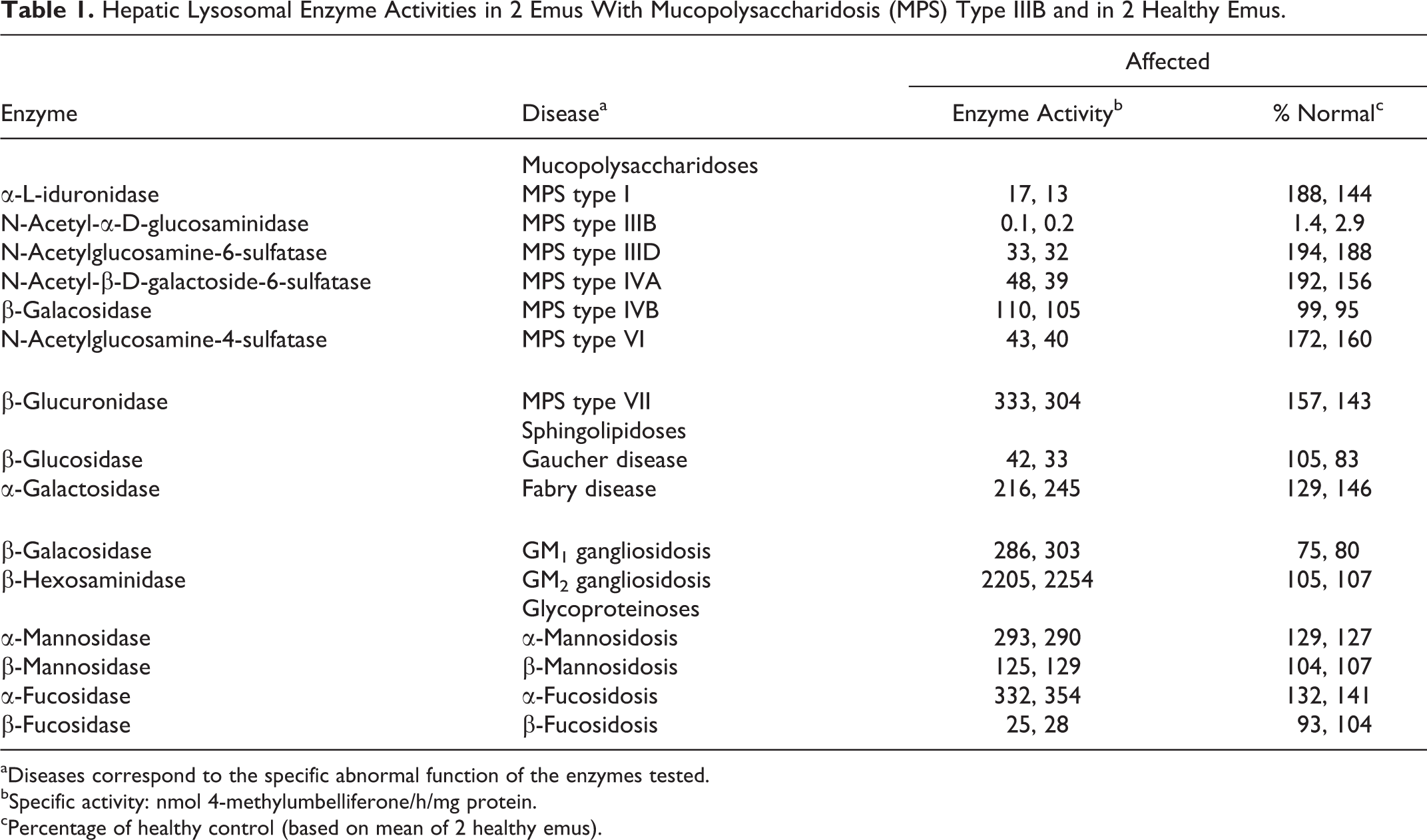
aDiseases correspond to the specific abnormal function of the enzymes tested.
bSpecific activity: nmol 4-methylumbelliferone/h/mg protein.
cPercentage of healthy control (based on mean of 2 healthy emus).
Serum NAGLU activity from 6 normal emus was 17.9 ± 3.7 nmol 4-MU/h/ml serum (n = 6). Serum NAGLU activities from the 2 families that had produced affected emus were very low, intermediate, or near normal (Fig. 11). The asymptomatic parents of affected emus had intermediate serum NAGLU activities, and affected offspring had low (<7%) NAGLU activity.
NAGLU genotyping
All emus in this study were genotyped for the previously described NAGLU mutation and the results were completely concordant. Clinically and pathologically affected emus with low serum NAGLU activity (n = 3) were homozygous for the mutant allele, and those with intermediate serum NAGLU activity (n = 7) were heterozygous for the mutant allele but clinically healthy. Those healthy emus with normal serum NAGLU activity (n = 8) were homozygous for the normal NAGLU allele.
Discussion
The age of the birds, the progressive neurological signs, and the light and electron microscopic features were consistent with a diagnosis of lysosomal storage disease. While the vacuolation of neurovisceral tissue initially suggested a gangliosidosis, biochemical studies revealed the presence of HS and a defective NAGLU enzyme, confirming an analogous disorder to the human Sanfilippo syndrome type B or MPS type IIIB.
The combined pathological and biochemical examination extends the initial description of this storage disease in emus. 4,5,31 Kim et al 31 described similar lesions in the brain, liver, spinal cord, and spleen of a 6-month-old male emu with lethargy and ataxia, but no biochemical assays were performed, so the enzymatic defect was not identified. Identifying the stored material and deficient enzymatic activity is required for the precise diagnosis of LSD and for distinguishing primary or secondary accumulation of storage material. Gangliosides can be stored as a primary substrate in GM1 and GM2 gangliosidosis. Alternatively, gangliosides can accumulate secondarily in MPS types I and III, 53 as seen in the affected emus here and in dogs previously. 15 Bermudez et al 4,5 described a hereditable gangliosidosis in the progeny of 2 emu breeding pairs: affected emus had increased amounts of gangliosides GM1 and GM3 and also contained ganglioside GM2 in the brain, normally undetectable in healthy emus. The occurrence of gangliosidosis was excluded by detecting normal hexosaminidase activity in the brains and only partially deficient activity of acid β-galactosidase. 5 Further characterization of the same animals suggested a possible sphingolipidosis due to the sphingolipid activator protein (SAP) deficiency, 17 although the biochemical mechanism of this LSD was never elucidated. Thus, the biochemical analyses presented here and the published molecular studies 1 appear to resolve the uncertainties surrounding those previously described storage diseases as an autosomal recessively inherited MPS type IIIB or Sanfilippo syndrome type B in emus.
In our cases, sudden death and severe hemorrhage from ruptured liver and other organs were unique presentations for affected emus. This condition has not been previously reported in any storage disease, the pathogenesis of which has yet to be elucidated. Abdominal and hepatic hemorrhages in these emus are most likely associated with spontaneous liver rupture secondary to hepatic lipidosis, while the other hemorrhagic lesions are difficult to explain. MPS may cause lesions in blood vessels, 11 as demonstrated by the cerebral vasculopathy and recurrent subdural hematoma described in a case of human MPS type IIIB: large amounts of GAGs in the intima and media as well as degenerative changes with mineralization of the internal elastic lamina of arteries have been reported. 2 However, histological examination of subcutis and skeletal muscle in our cases failed to reveal any major lesions. The same bleeding problems were described by Bermudez et al, 4,5 further suggesting those emus had MPS type IIIB. A possible explanation is a decreased production of coagulation factors secondary to liver disease, but this has not been investigated. Subcutaneous and muscular hemorrhages could also derive from trauma, due to severe weakness and loss of balance. Finally, the coagulopathy could be caused by elevated levels of heparan sulfate in the blood, as occurs in children with Sanfilippo syndrome 8 but not reported in affected dogs. Since most of the affected emus in the study experienced hemorrhages, this species potentially can be considered a model to study the increased bleeding tendency in Sanfilippo syndrome.
The severe progressive cerebellar signs, including ataxia, seen in some young affected birds are characteristic features of other species with MPS type IIIB, including dogs and humans. In the latter, after a symptom-free interval during the first months of life, a slow onset of mental retardation becomes apparent between 1 and 4 years of age. This is followed by severe behavioral problems and progressive intellectual decline around 3 to 4 years. 49 In Schipperke dogs, ataxia remains unnoticed until 2 to 3 years of age and progresses to severe incoordination and inability to stand and see by 5 to 6 years. 15 The different clinical phenotypes and disease course may be explained by differences in specific NAGLU mutations, residual NAGLU activity levels, species-specific effects on ganglioside, and HS accumulation and their effects on neuronal function.
The mechanism by which heparan sulfate storage leads to CNS degeneration is still unknown. It has been proposed that some of the CNS lesions observed in MPS type III are due to the secondary lysosomal accumulation of GM2 and GM3 gangliosides, resulting from the HS-induced interference with neuraminidase activity. 29 Gangliosides appear to stimulate meganeurites and neurite sprouting, and the presence of new neurites and their synapses apparently plays a role in the CNS dysfunction of these diseases. 51,53 Storage of gangliosides in the CNS has been shown to disrupt Ca2+ uptake by the rough endoplasmic reticulum Ca2+-ATPase, a process that is critical for proper neuronal function and survival. 42 Glycosaminoglycan storage abrogates autophagy, 44 causing severe neuronal lesions. 33 Inflammation, apoptosis, and oxidative stress have been reported to be involved in neurodegeneration in knock-out murine models of MPS type IIIB. 50 In addition to gangliosides, sequestration of cholesterol in the cell bodies of neurons and glia has been observed in Sanfilippo syndrome, causing aberrant lipid distribution throughout the cell that may disrupt membrane dynamics and intraendosomal transport. 52
The same vacuolation of neurons in the central 4,5,31 and peripheral nervous system, 5 retinal ganglion cells, 5 and renal epithelial cells 4 as well as infiltration of foamy macrophages in liver, spleen, lung, and renal glomeruli 4,5,31 also has been reported in emus assumed to have gangliosidosis. However, our study also describes extensive vacuolation in the autonomic ganglia in many organs (heart, lung, kidney, adrenal gland, ear) and infiltration of foamy macrophages in the aorta, intestine, pancreas, and thyroid gland. The foamy macrophages in a wide variety of tissues probably reflect uptake of HS from intercellular spaces and blood. These cells are presumed to contain soluble GAGs leaching during the preparation of histopathological sections, although in some, there was still some slightly PAS-positive material seen. The severe vacuolation of renal tubules may indicate high circulating levels of HS, which is excreted through the glomerular tufts to be reabsorbed in the tubules and ducts, rather than GAGs derived in situ from deranged catabolism.
In our cases, lysosomal storage was heterogeneous, ranging from clear vacuoles, such as those found in macrophages, to discrete round cytoplasmic inclusions of variable density with PAS staining in neuronal cells. The variable LFB and PAS staining of the vacuoles is further evidence of heterogeneity of storage material. Ganglioside oligosaccharides 10 and HS stain PAS positively, but in practice, results can be very variable. 3 The presence of LFB- and/or PAS-staining neurons in the CNS indicates storage of polar lipids and carbohydrate-containing vicinal glycal groups and saturated lipids, respectively. 34,40,41 These staining properties are consistent with the storage of GM2 and GM3 gangliosides, as demonstrated in MPS type IIIB in humans and other animals. 15,19,35
The detection of lipid vacuoles within the cytoplasm of hepatocytes (hepatic lipidosis) is consistent with previous reports, both in MPS type III 25,43 and other types of mucopolysaccharidosis, such as MPS types I and VI. 7,43 This fatty change is considered secondary to the metabolic derangement and cell injury caused by the accumulation of GAGs.
Three different types of inclusions have been described in our study by means of TEM, reflecting the different storage material accumulating within the liver, brain, and eye. Many hepatocellular inclusions contained prominent electron-dense bodies, similar to those previously described as GAG inclusions, 22,43,48 while the multilaminar membranous bodies have been shown to contain gangliosides that accumulate as secondary products in MPS. 9,19 In the retina, a mixture of both GAGs and gangliosides could represent a third type of inclusion consisting of a central core surrounded by membranous whorls.
Finally, the comparison of NAGLU activity among controls, homozygote-affected emus, and heterozygote parents confirms an autosomal recessive mode of inheritance of this disease in emus, as in most LSDs. Aronovich et al 1 described a homozygous NAGLU mutation in MPS type IIIB–affected emus characterized by a 2-bp deletion with elongation past the original stop codon to produce NAGLU with an extra 37 amino acids as well as 387 altered amino acids. This deletion occurred in all affected emus tested in this study, and mutations with a similar effect have been documented in humans with a severe phenotype. 54
In conclusion, emus have a disease similar to that seen in humans and dogs with Sanfilippo syndrome type B, making these animals potentially valuable for the investigation of disease pathogenesis and the testing of therapies, but an emu colony has not been developed. The current frequency of this inborn error of metabolism in the emu population remains unknown.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.