
Abstract
During the 20th century, as drug products were being developed to treat both known and emerging human diseases and conditions, determining the safety of these new chemicals became of increasing importance and necessity. For a time, the safety of use in human populations was of question, let alone whether the drug product was truly effective. As such, US and international regulatory agencies have played a major role in establishing standardized testing to evaluate the safety and efficacy of drug products. Pharmacologic and toxicologic evaluation of a new drug in animals is an important part of the pharmaceutical development process prior to its first-time use in humans, as well as its potential chronic use in affected populations. Just as both science and technology have evolved over the past century and further, so have the guidelines that have been put forth to adequately and efficiently evaluate the toxicity of new drugs and their subsequent safety in humans. This review summarizes the historical highlights of the conduct of drug safety evaluations in animals, particularly with regard to chronic toxicity and carcinogenicity assessments, and how we have progressed to our current standards and protocols to ensure safe use of drug products in human populations.
Older Chronology
Early in the 20th century, Katsusaburo Yamagiwa first proposed that chronic irritation could cause precancerous alteration in previously normal epithelium. 55 Soon after, in 1918, and nearly 100 years ago, the collaborative work of Yamagiwa and Koichi Ichikawa led to the first reports on experimental carcinogenesis: painting the inner surface of the rabbit ear with coal tar, now an over-the-counter pharmaceutical for psoriasis.54,55 Later, in 1929, studies involving experimental, multistep, dermal carcinogenesis in mice using benzpyrene, croton oil, coal tar, and mustard gas were first published by Isaac Berenblum, then a Riley-Smith Research Fellow in the Department of Experimental Biology & Cancer Research, University of Leeds. 7 Berenblum proved to be highly influential in the developing field of experimental carcinogenesis. In 1941, he demonstrated through experimental research that carcinogenesis induced by chemicals involves 3 separate and independent processes: initiation, promotion, and latency. 6 Berenblum also observed that every carcinogen that produces a tumor at the site of application or injection is an irritant, in the sense that it induces a continued state of reparative hyperplasia. 5 He further indicated that in all cases in which sufficiently accurate observations can be made, it is seen that the primary tumor is preceded by a stage of hyperplasia. Berenblum concluded that although hyperplasia is an essential precursor of neoplasia, only some and not all irritants are carcinogenic. Accordingly, preneoplastic hyperplasia must be a specific type and biologically distinct from ordinary reparative hyperplasia. Berenblum continued to work and publish in the field of carcinogenesis, primarily at the Weizmann Institute of Science in Israel, until his retirement in 1971.
In the United States, with the passage of the Food and Drug Act of 1906, the Food and Drug Administration (FDA; then known as the Bureau of Chemistry under the US Department of Agriculture) was charged with the responsibility for preventing the adulteration or misbranding of foods and drugs that were marketed to the public. 43 Beginning in 1938, the Federal Food, Drug, and Cosmetic Act gave regulatory powers to the FDA, requiring, among other things, that new drugs be clinically tested and proven safe prior to being sold. The FDA offered guidelines for such studies, but there were no designated standards.26,27,48 The FDA first published guidance for industry for assessing the toxicity of chemicals in food in 1949. 26 This guidance was referred to as the “black book” and included a contribution by O. Garth Fitzhugh on the subject of long-term studies and their design. Fitzhugh suggested that for long-term feeding studies, 2 species should be investigated: the albino rat would be studied for a lifetime of about 2 years, and a nonrodent second species (dogs or monkeys) would be studied for at least 1 year. Dose selection for these long-term studies would be based on results of subacute studies. Four groups of at least 10 animals of each sex were proposed: (1) a dietary control group, (2) a group fed a diet containing 100 times the amount of the substance proposed for use in food, (3) a group fed a diet containing the highest tolerated amount of the substance, and (4) a group given an intermediate dosage. Biochemical and hematology evaluations were to be made at 3-month intervals during the chronic study. At the end of the study, autopsies were to be performed, along with weighing of the principle organs and preservation of tissues for microscopic examination. The pathology evaluation was described in further detail in the black book by Arthur Nelson, indicating that tissues to be evaluated included lung, heart, spleen, pancreas, gall bladder, lymph nodes, stomach, small intestine, colon, kidney, adrenal, urinary bladder, testis or ovary, prostate or uterus, thyroid, parathyroid, submaxillary salivary gland, 4 levels of brain, hypophysis, bone, bone marrow, and voluntary muscle.
Lehman and coworkers 27 updated the FDA guidance in 1955 to include sections pertaining to drugs and recommendations for toxicity studies intended to support marketing applications. These studies included acute, subacute, and chronic toxicity testing, with chronic toxicology studies having a suggested duration/species of 2 years in the rat and 1 year in the dog. At this time, designated carcinogenicity studies had not been established; therefore, the carcinogenic potential of the drug product was assessed in the chronic toxicology study. In a section of these guidelines that addressed nonclinical carcinogenicity assessment, contributing author Anne Bourke referred to some of the variables in carcinogenicity studies, including genetic predisposition to spontaneous tumors, composition of the basal diet, and the suspending or diluting fluid (vehicle) of the substance to be tested. Most importantly, she noted that the conclusions for one route/species could not necessarily be extrapolated across routes or species, writing that “positive results in an animal test can be taken as creating a suspicion that the chemical under study may be carcinogenic for man, but do not prove it to be so.” Arthur Nelson was also a contributing author to these guidelines and expanded upon the pathology evaluation in long-term studies, with suggestions to improve the quality.
In 1958, when the US government passed food additives amendments requiring manufacturers to establish safety and to eliminate additives that were demonstrated to cause cancer, the FDA’s focus was on food safety. This changed in 1962, when the congressional Kefauver-Harris amendment to the Food, Drug, and Cosmetic Act was enacted to promote drug safety, shifting the burden of proof of clinical safety of drugs to drug manufacturers. For the first time, drug manufacturers had to prove that their products were both safe and effective before they could be sold. Subsequently, guidelines for toxicity tests for all drugs (known as the Lehman Guidelines) were written by Arnold Lehman, director of the Division of Pharmacology of the FDA, to aid the pharmaceutical industry in complying with the new law. Rats, dogs (beagles), and rabbits were the primary species for testing at this time, with chronic toxicology tests conducted for 1 year to 18 months depending on the species. 57 Around the same time as the Kefauver-Harris amendment, in 1962, the National Cancer Institute (NCI) Carcinogenesis Screening Program was initiated.
Development of Standardized Protocols
Early efforts to develop standardized carcinogenicity protocols were begun in the 1960s, when NCI scientists John and Elizabeth Weisburger began revising standardized systemic carcinogenicity protocols based on FDA protocols.45,46 Their design is the basis for the current design for 2-year carcinogenicity studies in rodents. The importance of the purity and stability of the test chemical, selection of an appropriate test species, standardization of animal maintenance and environmental control (including temperature, humidity, hours of light, bedding, airflow, water, and diet), and issues related to various routes of administration and dose selection were all addressed. The duration of studies (eg, 24 months), in-life observation, suggestions for necropsies, tissue fixation, diagnoses of lesions by a highly trained person, the importance of detailed record keeping, and statistical analysis were all considered. As a demonstration of how the testing evolved early on, in 1961, an NCI carcinogen screening test of a given chemical performed in one species took as little as 8 months and cost about $10,000 to $15,000. Ten years later (1972), a more extensive test in 2 species with larger numbers of animals required about 30 months and cost about $75,000. 47 By 2009, costs for carcinogenicity testing in 2 species were in the range of $2-4 million. 2
Beginning in 1968, drug package inserts were required for newly approved drugs, including a section discussing carcinogenesis. The following year, data elements to be captured for carcinogenicity studies were described by Berenblum and included descriptive information on the chemicals, animals, experimental design, survival, body weight, and individual pathologic results, as recommended by the International Union Against Cancer. 4 Also during this time, the FDA had established recommendations for chronic toxicity and carcinogenicity assessment for pharmaceuticals seeking approval for marketing. Chronic toxicology studies were initially required in 2 species: 18-month rat and 12-month dog (and sometimes monkeys). Later, a 12-month chronic rat toxicity study was accepted in place of the 18-month chronic rat study, provided a 2-year mouse carcinogenicity study was conducted.11,15
It was noted that the selected duration of chronic studies was influenced by toxicities elicited by a number of anticonvulsants, analgesics, hypercholesterolemic agents, and tricyclic antidepressants, which were being manifest only between 6 and 12 months of treatment. Furthermore, in the early 1970s, with the initiation of the National Cancer Program and its Carcinogenesis Testing Program, the NCI was asked by the FDA to conduct carcinogenicity studies in rats and mice for some older drugs. An electronic data capture system for these NCI carcinogenicity studies, the carcinogenesis bioassay data system (CBDS), was also developed during this time, 30 and an NCI pathology working group was organized for peer review. The design of these early NCI studies was such that one set of 20 controls was used for several drug/chemical studies conducted in one room, with 50 animals used for each of the low- and high-dose groups for each unique drug/chemical.
Protocols for carcinogenicity studies conducted by the NCI Carcinogenesis Testing Program were first standardized in 1976. 33 The design was changed in that the number of controls was increased to 50, and each study was conducted in a separate room, with animals group-housed and having their own set of control animals. In 1978, the Carcinogenesis Testing Program was transferred from the NCI to the National Toxicology Program (NTP). For many years, animal dose administration in nearly all NCI/NTP gavage carcinogenicity studies, including those for drugs, was 5 days per week, even though humans would be prescribed the drugs for 7 days per week. For pharmaceuticals today, the frequency of administration usually mimics the frequency of administration to humans.
Standardized carcinogenicity protocols were published by the Organization for Economic Co-operation and Development in 1981 and then described in the 1982 FDA Redbook. These protocols specified the use of 3 dose groups, the highest being a maximum tolerated dose (MTD). The definition of MTD used by the NCI and the NTP was also used for many years by the FDA and was defined as “the highest dose of a test agent used during the chronic study that can be predicted not to alter the animals’ normal longevity from effects other than carcinogenicity.” 33 Other interpretations vary as to exactly what constitutes an MTD, with the various definitions discussed in ICHS1C(R2). 19 The MTD is now considered by the Center for Drug Evaluation and Research (CDER)/FDA to also include severe alterations to homeostasis or other alterations that might interfere with interpretation of the studies. By the mid-1980s, carcinogenicity studies of new drugs conducted by a drug sponsor for the FDA included 3 dose groups of at least 50 animals for rats and mice. In 1987, CDER was established within the FDA (from the Center for Drugs and Biologics), and soon after, the Carcinogenicity Assessment Committee (CAC) and executive CAC were formed to review carcinogenicity protocols and results to ensure consistency across the center. Protocols generally followed the NCI/NTP design, except using 3 dose groups and dosing that mimicked the clinical frequency of dosing. Although quality assurance procedures for pathology assessment were outlined by the NTP, 9 CDER/FDA has relied almost entirely on the sponsor for the pathology assessment, and only occasionally is an outside peer review requested. In contrast, individual animal data are submitted to CDER/FDA for statistical analysis by CDER statisticians so that statistical analysis can be standardized and applied to be the same for all carcinogenicity studies of drugs. 28
History of Carcinogenicity Studies With Oral Contraceptives
Oral contraceptives were first being developed and seeking FDA approval in the 1950s. At that time, there were no specific studies in place to assess the carcinogenicity of drug products since it was only 1962 when drug developers were required to demonstrate drug safety. The design of animal toxicity studies to support the clinical safety of the drug product was often left to the discretion of the sponsor, and even when guidelines were established in 1955, the carcinogenic potential of the drug product was typically assessed in the chronic toxicology studies.
Enovid (norethynodrel and mestranol) was the first oral contraceptive approved by the FDA. Initially approved in 1957 to treat gynecologic disorders, it did not carry a contraceptive claim until 1960. More or less concurrently, new drug applications for other steroid contraceptive combinations were also being submitted for approval by the FDA. Oral contraceptives, however, were unique in that, unlike other drugs being submitted for approval, oral contraceptives were not developed to treat a medical condition or disease but were instead intended to be administered to a healthy, nondiseased population. Furthermore, these contraceptives had no established therapeutic advantage and would be taken for an extended period of time, potentially years. Nonclinical testing to support the safety of contraceptives during this time was generally in accordance with FDA requirements for orally administered drugs intended for unlimited periods of use.8,32
As mentioned above, in 1962, mainly in response to the thalidomide tragedy, the Kefauver-Harris amendment to the Food, Drug, and Cosmetic Act was enacted to establish formal safety and efficacy requirements for new drugs.8,43 Even after publication of the Lehman Guidelines, the safety of oral contraceptives was tested in the same way as other drug products, without any established provision for special testing of contraceptives. 8 Ultimately, guidelines specifically developed for the investigation of the safety of oral contraceptives were proposed and presented to the FDA Advisory Committee for Obstetrics and Gynecology (OB/GYN) for discussion in November 1965: “Current Criteria for Evaluation of Progestational Agents and Oral Contraceptives: Preclinical Investigations.”8,44 While these guidelines did not provide specified tests, they described pharmacologic and toxicologic investigations and pointed out areas unique to the pharmacologic and toxicologic evaluation of hormonal agents that warranted increased scrutiny. These included, among others (1) the possibility of hormonal stimulation by higher dose ranges; (2) for the final evaluation of safety and efficacy, the compound should be tested by the route proposed for clinical use; (3) components of a combination should be first assayed separately but in the form of intended therapeutic formulation at the time of final evaluation; (4) new compounds with even slight changes in chemical structure could not reference a close relationship to the original compound and be considered safe, but would need a complete experimental evaluation as a new compound; (5) adequate data to establish the absence of the teratogenic potential of the compound should be provided, in addition to the usual required toxicologic background data, prior to phase 1 or 2 clinical studies.
In 1966, the long-term safety of oral contraceptives in humans was brought into question based on findings that the investigational combination oral contraceptive MK-665 (ethynerone plus mestranol, 20:1 ratio) had produced mammary tumors in beagle dogs and mammary hyperplasia and suspected tumors in monkeys following 1 year of exposure.8,13 The FDA felt that further studies must be undertaken to ensure that these lesions did not occur with other marketed oral contraceptive products and therefore drew up further requirements for testing steroid contraceptives, submitting them again to the FDA OB/GYN Advisory Committee for expert advice and review. Letters were sent from the FDA to manufacturers of steroid contraceptives in the fall of 1967 alerting them to one of the new requirements: initiation of long-term toxicity studies in primates (10 years) and beagle dogs (7 years) for hormonal contraceptives.8,13 The list of new requirements was then published in 1968, thus setting forth the establishment of the safety of oral contraceptive products in development. The new requirements included (1) a 1-year toxicity study in the rodent and dog (with studies in the monkey recommended but not required) prior to the initial clinical evaluation of a contraceptive product, (2) initiation of a 7-year study in the dog and a 10-year study in the monkey prior to beginning the phase 3 clinical trial for new estrogens or progestogens, and (3) completion of 2-year studies in the rat, dog, and monkey with results submitted with the marketing application. 15 In late 1969, this policy changed slightly in that completion of 2-year studies in rat, dog, and monkey were required prior to initiating phase 3 clinical trials (Table 1). 14
Recommended Animal Toxicity Studies for Contraceptives, Estrogens, and Progestogens: US Food and Drug Administration, November 1969.a
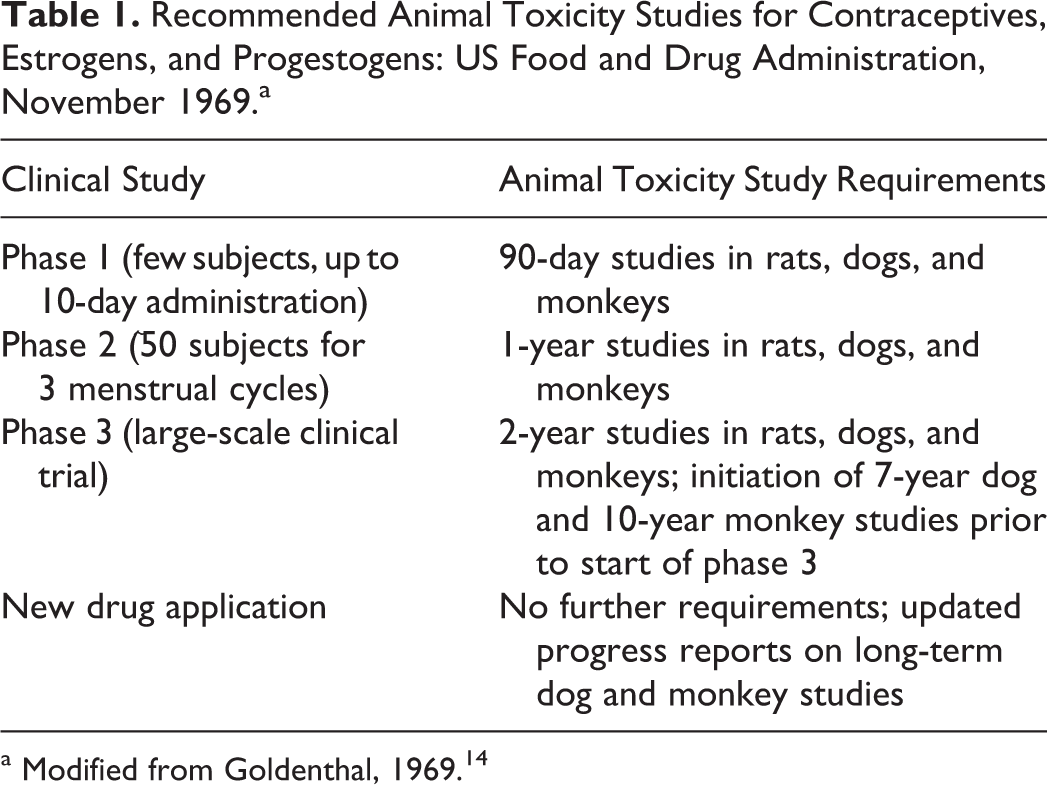
a Modified from Goldenthal, 1969. 14
The details of the design of the 7- and 10-year long-term studies have been discussed by Berliner and Finkel and Berliner.8,12 The choice of dog (beagle) and monkey (rhesus) as species was based on the history with the contraceptive component, MK-665 (ethynerone, which caused mammary gland nodules in dogs), experience of use of both species in toxicology tests, and the ready availability of the animals. At the time, the rodent did not appear to be a representative animal model for oral contraceptive products, even though it was the species of choice for carcinogenicity assays with other drugs. 8 The recommended doses in the 7-year dog and 10-year monkey studies were 2, 10, and 25 times the anticipated human dose for the dog and 2, 10, and 50 times the anticipated human dose for the monkey, both based on mg/kg body weight of the combination (both progestogen and estrogen were elevated at each multiple, maintaining the same ratio).8,12,25 Those sponsors who were developing products with more than one ratio of the same progestogen and estrogen were permitted to choose the most popular ratio and use this in the carcinogenicity study, rather than test all ratios. Pharmacologic evaluations were to be done to ensure that there were no marked endocrinological differences between the ratio of the tested formulation and the others available. 8 The 7- and 10-year duration of the studies represented half the life span in each animal, since human females could choose to take oral contraceptive products for half of their lifetime. With respect to the number of animals to be studied in each assessment, 12 animals per dose group were recommended as a minimum to be sufficient for statistical significance of results, even though many studies used 16 to 20 animals per dose group. In addition, drug administration was recommended to be by the same route and schedule as would be used in the clinical population (eg, oral; 21 days with treatment and 7 days without treatment, to mimic the clinical regimen). Overall, the studies were designed with the hope of determining (1) general toxicity and organ-directed toxicity not related to hormonal and metabolic effects, (2) toxicity related to hormonal effects, and (3) toxicity related to metabolic effects. 56
Results of these long-term studies started to flush out the effects of the various progestogens and estrogens being used in oral contraceptive products. In 1973, Finkel and Berliner 12 summarized the results of 8 studies that were then in progress investigating the carcinogenicity of the various oral contraceptive formulations. At the time, it was found that (1) if the progestogen component was a testosterone derivative (eg, norethindrone, norgestrel), it was less likely to induce tumors than progesterone derivatives used as the progestogen component (eg, chlormadinone, medroxyprogesterone); (2) progesterone analogues (except megestrol and ethynerone) caused a high incidence of pyometra and death; and (3) estrogen components were not tumorigenic.12,25 The results also raised doubts as to whether the dog was an appropriate test species due to differences in estrus cyclicity from humans, the potential differences in steroid metabolism between dogs and humans, and the reported incidence of spontaneous mammary tumors in the dog. Interpretation of the lesions in both dogs and rhesus monkeys had also been controversial. Although malignant and benign nodules were found in dogs following long-term treatment, monkey studies were predominantly negative. At the time, mammary carcinoma was considered to be uncommon in monkeys, and the background rate of mammary carcinoma in humans was between that of dogs and monkeys. Participants in a Consultation Conference convened by the Human Reproduction Unit of the World Health Organization (WHO) in 1976 were asked to deliberate on the use and acceptability of the beagle in the long-term studies required by the FDA for steroid contraceptives. The group concluded that “the beagle is a reasonably good breed for carcinogenesis testing, e.g. it is not refractory to the development of mammary tumors, yet not as susceptible to mammary tumors that they would frequently occur by chance alone” (V. R. Berliner, 1976, personal communication). Following these discussions, the long-term carcinogenicity studies in the dog continued.
The guidelines for long-term carcinogenicity studies of oral contraceptives remained in place until the mid-1980s. In 1984, the International Conference of Population put forth a request to modernize and update the official requirements for the preclinical and clinical assessment of new fertility-regulating agents. 51 In response to this request, the WHO convened an international meeting and produced “Guidelines for the Toxicological and Clinical Assessment and Post-Registration Surveillance of Steroidal Contraceptive Drugs”50,51 based on feedback from international experts and WHO scientists. With regard to carcinogenicity testing, the WHO guidelines eliminated the 7-year dog and 10-year monkey studies, substituting the use of a 2-year rat or an 18-month mouse study to assess carcinogenicity of new contraceptive products. The FDA then convened a meeting of its Fertility and Maternal Health Drugs Advisory Committee to discuss these guidelines and consider whether they were acceptable for adoption by the FDA. 42 Based on input and recommendations from WHO scientists and consultant toxicologists, and the advice of the Advisory Committee, the FDA chose to revise their requirements for testing steroidal contraceptives to better reflect current scientific opinion and conform more closely to the new WHO guidelines. However, the FDA also chose to modify some of the WHO guidelines for toxicity and carcinogenicity testing to better coordinate with then-current FDA guidance. A summary of the previous and newly revised FDA requirements for carcinogenicity testing for drug products and contraceptive drug products based on the 1987 WHO guidelines is described in Table 2.
Summary of Past and Present Guidelines to Evaluate Carcinogenicity of Contraceptive Drugs (as of December 1987).a

Abbreviations: FDA, US Food and Drug Administration; WHO, World Health Organization.
b Interim period until the WHO epidemiological study of depot-medroxyprogesterone was completed.
At the time these revised guidelines were accepted by the FDA, there were epidemiologic studies in progress by the WHO that were investigating the link between both combined oral contraceptives in general and depot-medroxyprogesterone acetate (DMPA), and breast cancer.49,52,53 Although the FDA agreed with the WHO that the carcinogenicity studies in dogs and monkeys should be discontinued, pending the results of the WHO investigation, the FDA retained a required carcinogenicity study in dogs but reduced the duration to 3 years. The reduced duration was based on evidence that all steroids that produced neoplasms in beagles did so within 3 years’ time. 25 The final results of the WHO epidemiological studies showed that there was no difference between DMPA and marketed oral contraceptives in overall relative risk for breast cancer in humans.49,53 Hence, it was concluded that the carcinogenicity found in beagles did not predict the effects in the human population. As a result, the FDA discontinued the requirement of long-term carcinogenicity studies of contraceptive steroids in dogs. 24 Another change made by the FDA to align carcinogenicity studies in contraceptive steroids with other drug products was to increase the duration of the mouse carcinogenicity study from 18 months to 2 years (S. Sobel, 1991, US FDA communication). 24 Current FDA guidance still recommends 2-year carcinogenicity studies in both mouse and rat for novel contraceptive products under development. Per ICHS1B, 18 a 6-month transgenic mouse study could be conducted in place of the 2-year study in mice.
History of Short-term Carcinogenicity Bioassays (or Alternative Methods of Carcinogenicity Testing) for Pharmaceuticals
In 1997, the FDA agreed to ICHS1B “Testing for the Carcinogenicity of Pharmaceuticals,” which allowed for the use of transgenic models in the evaluation of drugs. 18 The first results from such studies were submitted in 1998. In 2001, results of an extensive, collaborative review coordinated by the International Life Sciences Institute/Health and Environmental Sciences Institute of some alternative carcinogenicity methods in mice were published, encompassing an entire issue of Toxicologic Pathology. 40 Methods reviewed included the P53+/– model, the Tg.AC model, the TgHras2 model, the XPA and XPA/P53+/– model, and the neonatal model. After publication of this supplement, the number of carcinogenicity protocols for alternative models submitted to CDER/FDA increased dramatically.
Initially, most of the protocols were for the P53+/– mouse model (25 animals per group) for equivocally or clearly genotoxic compounds and the Tg.AC mouse model (25 animals per group) for dermal products. Because all of the P53+/– studies after phenolphthalein were negative, the criteria for using that model were revised so that the drug had to be clearly genotoxic in the Ames test before conducting a P53+/– study. The early experience of CDER/FDA with the alternative carcinogenicity models was described by Sistare and Jacobs in 2003, 38 at which time most alternative assay protocols were for the P53+/– and Tg.AC mice. After many dermatologic vehicles were found to be clearly positive in the Tg.AC model, and because the model cannot distinguish between promotion and de novo carcinogenicity, use of this assay diminished to none by 2012. Once the TgHras2 mice (25 animals per group) became commercially available in the United States, use of these mice for either genotoxic or nongenotoxic drugs increased to the point that the number of protocols submitted for the TgHras2 model exceeded that for all other models. As more results for drugs have been submitted, confidence in the TgHras2 model has increased; ovarian neoplasms for selective estrogen receptor modulators have been detected, as have duodenal neoplasms for a tyrosine kinase inhibitor causing such neoplasms in rats. The assay is generally considered acceptable for all systemically administered drugs. It is noted that the power to detect hemangiosarcomas or pulmonary neoplasms is reduced in the TgHras2 mice because of the high and variable background incidence of these neoplasms in these mice.
The International Conference on Harmonisation (ICH) and Technical Requirements for Registration of Pharmaceuticals for Human Use (Dose Selection, Number of Dose Groups, Species and Strain, Duration)
The ICH discussions among the European Union (EU), Japan, and the United States began in the early 1990s to harmonize various issues related to carcinogenicity studies. Several different finalized ICH documents discuss the need for carcinogenicity studies and dose selection.17–19 Although the FDA Redbook 2000 and FDA Redbook 2006-2007 contained outlined carcinogenicity protocols, CDER/FDA has deviated and followed ICH guidelines with regard to dose selection in particular.
Dose Selection
Dose selection for chronic, noncarcinogenicity studies has not been addressed by ICH, except that a limit high dose of 50-fold the human exposure may be used in the absence of an MTD or maximum feasible dose. 16 For 2-year carcinogenicity studies, the ICH guidance allows considerations other than the MTD for the high-dose selection. These alternatives were considered acceptable according to an international agreement, 19 with the 6 generally acceptable criteria for selection of the high dose for orally administered pharmaceuticals being (1) the MTD, (2) a minimum of a 25-fold area under the plasma concentration-time curve (AUC) ratio (rodent: human) of systemic exposure for nongenotoxic drugs, (3) dose-limiting pharmacodynamic effects, (4) saturation of absorption, (5) maximum feasible dose, and (6) a limit dose. Regarding the use of dose-limiting pharmacodynamic effects, the ICHM3(R2) guidance specifically states that “the high dose selected should produce a pharmacodynamic response in dosed animals of such magnitude as would preclude further dose escalation, but not compromise the validity of the study.” 16 Examples include hypotension (adverse cardiac effects) and inhibition of blood clotting (because of the risk of spontaneous bleeding). Availability of systemic AUC measurements in both animals and humans allowed the use of the internal dose for making dose selection and dose-spacing decisions. Thus, spacing of doses for oral studies is based on the systemic AUC, not the nominal dose. For most drugs, dosing is most commonly conducted via gavage.
Number of dose groups
Studies submitted to the FDA may include untreated controls, vehicle controls, and 3 dose groups. Untreated controls may be used when a proposed vehicle has not previously been studied in 2-year studies. Since 2000, a number of studies submitted to CDER/FDA have included dual (identical) control groups. Several cases of differences in neoplasm incidences, such as 11/60 versus 1/60 for the dual controls have been observed, which has informed on the variability of the background incidences, even after randomization. Changes in background incidence for various neoplasms may vary from supplier to supplier, from testing lab to testing lab, and over time. As for number of animals per group, once 2-year studies were expected, many sponsors increased the group sizes from 50 to 60-70 animals, in case of poor survival.
Controls
Most carcinogenicity studies of drugs involve gavage administration with varying vehicles. When the vehicle control is not water or carboxymethylcellulose, and the testing laboratory does not have 2-year toxicity data on the vehicle, a water or saline gavage vehicle control may be recommended in addition to the proposed vehicle. In some cases, toxicity of vehicles has confounded interpretation of carcinogenicity studies.
Species and strain
For chronic toxicology studies, the dog has generally been the preferred nonrodent species for pharmaceutical testing. However, minipigs have become a preferred species for dermally applied drugs because of the closer similarity of minipig skin to human skin. For carcinogenicity studies, although the NCI/NTP initially chose to use the F344 rat and B6C3F1 mice strains with a single feed and animal source, by the late 1970s, the choice of rat and mouse strains for pharmaceutical testing was left to drug developers. Initially, Sprague-Dawley rats and CD-1 mice were the primary species chosen for these studies. However, some studies conducted for the FDA by the NTP and studies conducted at the National Center for Toxicological Research were conducted in F344 rats and B6C3F1 mice. Over time, more and more rat studies have been conducted using the Wistar strain. Because the strain and source of the animals is left up to the drug sponsor, this means that studies conducted by sponsors for the FDA have used varying strains of rats and mice, and from various sources, resulting in very different background rates for neoplasms across studies (eg, there could be US, European, or Asian sources of animals and feed).
Transgenic mice, as discussed earlier, have also become important strains for use in carcinogenicity studies. Between 1998 and 2012, CDER had received approximately 240 protocols for transgenic studies versus 1500 protocols for traditional carcinogenicity studies. In 2011, 40% of the protocols for carcinogenicity studies in mice were for TgHras2 mice.
Duration
For chronic studies, after an extensive data review at the FDA, and per the original ICHM3 guidance document, which was finalized in 1997, the duration of studies was reduced to 6 months for rodents and 9 months for nonrodents from the previous 1-year suggestion. At the time of the ICHM3 guidance revision in 2009, another data review (unpublished) of drugs and duration of studies was conducted by Japan, the EU, and the United States. The review for nonrodents included all available data sets for drugs, primarily for dogs, between 1999 and 2006. The results of this review by the ICHM3(R2) expert working group led to the conclusion to maintain the duration of studies at 6 months for rodents and 9 months for nonrodents, since there were sufficient examples of studies in nonrodents showing effects that changed the clinical course of development after 6 months. 16 For biologic compounds, however, a 6-month chronic study in a nonrodent may be sufficient for non-oncology indications.20,21
For carcinogenicity studies, 2 years for traditional rat and mouse protocols and 6 months for transgenic mouse protocols are the standard durations. However, for biologics, it is not always appropriate or possible to conduct traditional carcinogenicity studies, especially when rodents do not express the pharmacodynamic activity. ICH S6(R1) suggests that the sponsor propose to CDER/FDA how they intend to address the carcinogenicity potential of their biologic product, together with supporting information and rationale. 20 The proposal may or may not include a 6-month or a 2-year carcinogenicity study. The proposals are discussed with the executive CAC and interested CDER division representatives. In 2011, more than a dozen such proposals were reviewed by CDER.
Analysis of Rodent Carcinogenicity Studies
It is desirable to have histopathologic diagnoses made according to standardized criteria and nomenclature. CDER/FDA does not specify what criteria to follow, so in the United States, drug sponsors generally follow the NTP criteria or the Society of Toxicologic Pathology criteria being used at the time. However, criteria used in other countries are not always supplied and may differ from those used in the United States. The international discordance should eventually be remedied by the INHAND Project (International Harmonization of Nomenclature and Diagnostic Criteria for Lesions in Rats and Mice), 22 which is a joint initiative of the Societies of Toxicologic Pathology from Europe (ESTP), Great Britain (BSTP), Japan (JSTP), and North America (STP) to develop an internationally accepted nomenclature for proliferative and nonproliferative lesions in laboratory animals.
A publication by McConnell et al 31 and the updated version by Brix et al 10 are generally followed for decisions on how to combine neoplasms for statistical analysis. Statistical analysis of neoplasm incidences has changed over time at CDER/FDA, as methods used by the NTP have evolved.29,37 All studies submitted to CDER/FDA are analyzed by CDER statisticians by the methods being used by CDER/FDA at the time. The Peto statistical analysis is still used despite issues of often not knowing whether the neoplasm of interest was lethal or incidental and the unreliable results obtained when the incidences are small. The poly-3 method of analysis is based on an analysis of spontaneous neoplasms in B6C3F1 mice and F344 rats in the NTP program and adjusts for survival differences but does not use time intervals or require knowledge of whether a neoplasm is fatal or incidental.3,34 Periodically, the statistical methods used in CDER are revised.
Historical background neoplasm incidences in studies submitted to CDER/FDA are difficult to use because of the variability of numerous factors: across time, various strains, animal sources across the world, and diets, to name a few. There was a short period during which a few laboratories in the United States used caloric-restricted diets, which resulted in lower incidences of background neoplasms, but no such studies have been received for the past 8 years.
Current Issues and Relevance to Humans
At present, it is not always clear which nonhuman model is the most appropriate for assessing chronic toxicity or carcinogenicity of pharmaceuticals. Sometimes normal animals do not possess the excess or deficiency that is characteristic of the disease model of treated patients and may also exhibit toxicities to excesses or deficiencies that humans would not display. For example, a drug intended to reduce blood pressure in hypertensive persons will cause dose-limiting hypotension and other adverse effects in normotensive animals. Likewise, a drug designed to reduce blood sugar will cause hypoglycemia in nondiabetic animals. Furthermore, animal models of the disease may not be robust, and for these reasons, human clinical data may best inform as to which nonhuman model is most appropriate in studying drug toxicity.
Most pharmaceuticals are not genotoxic, and since pharmacodynamic activity may be exaggerated in animals, this can thereby affect the relevance of carcinogenicity findings to humans. Thus, the mode of action may not generally be initiation (direct DNA reactivity), promotion, and progression in that order. For CDER/FDA, information such as pharmacology, receptor binding, P450 enzyme effects, human pharmacokinetics and toxicity, and other information are all considered and play a role in interpreting carcinogenicity studies.
Thus far, in vitro and in silico assays have not been particularly predictive for drug toxicity in animals or humans. Relevance to humans has increasingly been considered for positive carcinogenicity findings, and in vitro or ex vivo assays have sometimes been useful in understanding effects across species. The role of differences in genetic susceptibility, pathways affected by the drug, epigenetic effects, control of or levels of hormones, and receptor presence and density has increasingly been recognized in consideration of cross-species relevance.
The Future
In contrast to high-production-volume chemicals or environmental contaminants, humans are exposed to substantial doses of drugs, generally with intentional major perturbations to homeostasis. Many drugs negative for genotoxicity, but with positive carcinogenicity findings in animals, have such findings that are usually secondary to the pharmacologic effects or due to other epigenetic effects at doses relatively higher than those that humans receive. Such carcinogenicity findings rarely affect approval of a drug, as labeling of approved drugs has illustrated. 41
Because of deficiencies of animal carcinogenicity studies, 1 and based on some extensive data reviews, some proposals have been made by representatives of the pharmaceutical industry to the ICH for refining the criteria for when carcinogenicity may or may not be warranted for pharmaceuticals. 39 Alternative (unpublished) proposals have been submitted by other parties to the ICH as well. It is hoped that discussions will be fruitful.
Many have questioned the continued need for rodent carcinogenicity studies for pharmaceuticals and have asked whether aspects of the assessment model for evaluation of carcinogenicity for biologics 20 could be used for small molecules. Per ICHS6(R1), 20 a strategy for assessment of carcinogenicity potential could be based on a review of relevant data from a variety of sources. The data sources can include published data (eg, information from transgenic, knockout or animal disease models, human genetic diseases), information on class effects, detailed information on target biology, in vitro data, data from chronic toxicity studies, and/or clinical data. The product-specific assessment of carcinogenic potential is used to communicate risk and provide input to the risk management plan along with labeling proposals, clinical monitoring, postmarketing surveillance, or a combination of these approaches. In some cases, the available information can be considered sufficient to address carcinogenic potential and inform clinical risk without warranting additional nonclinical studies. For example, immunomodulators and growth factors pose a potential carcinogenic risk that can best be evaluated by postmarketing clinical surveillance rather than further nonclinical studies. Perhaps this approach can be extended to small molecules, in conjunction with some other considerations, such as those suggested by the Pharmaceutical Research and Manufacturers of America (PhRMA). 39
As time goes on, more and more carcinogenicity findings of drugs in rodents are not considered to be relevant to humans; however, the occasional clinically relevant finding of carcinogenicity in rodents does appear. Some have proposed using improved in vitro assays, ex vivo assays, and improved in silico assays in conjunction with what is known about pharmacologic activity, both on target and off target, and shorter-term toxicity, to make an assessment for initial labeling of drugs, with follow-up in humans. Better methods to assess systemic interactions and downstream effects without using animals would be needed before more reliance can be placed on in vitro assays. Given that hyperplasia at 3 or 6 months does not predict carcinogenicity in rodents at 2 years,23,39 it remains unclear how acute exposure in vitro can capture long-term effects. Rather than conduct actual carcinogenicity studies and report the findings in product labeling, the probability of adverse long-term effects could instead be described in the drug labeling, based on the entire knowledge base. Improved in vitro, ex vivo, in silico, or biomarker assays are needed before a completely revised approach is likely to generally become a reality. In the mid-term, perhaps a revision of, or alternative to, the PhRMA proposal, incorporating a few additional considerations such as pharmacologic or toxicologic mode of action, may reduce the number of rodent carcinogenicity studies conducted.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.