
Abstract
Neoplasia in both animals and humans results in part from lasting activation of tumor-promoting genes (“oncogenes”) or diminished function of genes responsible for preventing neoplastic induction (“tumor suppressor genes”). The concept of “genetic addiction” has emerged to indicate that neoplastic cells cannot maintain a malignant phenotype without sustained genotypic abnormalities related to aberrant activity of oncogene(s) and/or inactivity of tumor suppressor gene(s). Interestingly, some genetic abnormalities reliably produce distinct morphologic patterns that can be used as structural signatures indicating the presence of a specific molecular alteration. Examples of such consistent genetic/microanatomic pairings have been identified for mutated oncogenes, such as rising mucin-producing capacity with RAS overexpression, and mutated tumor suppressor genes—including PTEN eliciting cell hypertrophy, RB1 dictating neuroendocrine differentiation, and TRP53 encouraging sarcomatous transformation. Familiarity with the concept of genetic addiction, as well as the ability to recognize such regular genomic-phenotypic relationships, are of paramount importance for comparative pathologists who are engaged in phenotyping genetically engineered mice to help unravel genomic intricacies in both health and disease.
Keywords
Some Genetic Mutations Can Be “Seen” Under the Microscope
The concept that specific etiologic agents can yield consistent morphological changes is a traditional foundation of pathology. The classic example is infectious disease, where the basic patterns of microbe-induced inflammation are suppurative infiltrates for acute bacterial infections, granulomatous reactions for chronic bacterial and fungal infections, lymphocytic aggregates (often perivascular) for many viruses, and eosinophilic influx for metazoan parasites. In like manner, specific gene abnormalities in certain human cancers can be visualized by the pathologist as stereotypical structural changes in malignant tissues. For instance, in humans, the outcome of mutations to the BRCA1 gene, which encodes a tumor suppressor protein with DNA repair functions, is visible in the breast as medullary carcinomas and in the ovaries as serous carcinomas. Conversely, the loss of E-cadherin in human breast cancer results in a lobular pattern. 3,21,24,29,32,104,109 The recognition that certain genetic abnormalities are reliably connected to distinct patterns of morphological abnormalities represents an important new paradigm in cancer biology.
This recognition has been formalized during the past decade as the concept of “oncogene addiction.” This hypothesis, which emerged in the literature between 2000 and 2002, states that neoplasms depend on continued expression of one or more oncogenes for maintenance of the malignant phenotype. This premise is supported by findings in some inducible mouse models of human cancer in which tumor regression occurs after a particular oncogene has been “switched off.” 134 For example, persistent expression of c-Myc in murine hematopoietic lineages results in T cell and myeloid leukemias, whereas events that inactivate c-Myc in leukemia cells lead to a rapid decline in cell division in conjunction with a pronounced rise in apoptosis. 30 The similar concept of “tumor suppressor gene addiction” has been developed as an explanation for the phenomenon that most tumors require lasting inactivation of one or more tumor suppressor genes to ensure continuation of the malignant phenotype. The basis of this concept is that neoplasms will regress or disappear once the function of the missing tumor suppressor gene is restored. The idea is bolstered by studies in mouse cancer models where regression of hepatocellular carcinomas, lymphomas, and sarcomas may be achieved upon restoration of TRP53 function. 130,138 A recent review has revisited the concept of “gene addiction” and broadened its meaning to include both oncogene addiction and nononcogene addiction (NOA). 74 NOA-related genes do not necessarily initiate cancer but instead play critical roles in cancer progression and biological behavior because they encode various molecules involved in almost all critical signaling pathways. Because NOA genes are usually downregulated or lost in cancer, they too fit under the broad umbrella of “tumor suppressor genes.”
As the catalog of new mouse models of human cancer grows, the link between certain oncogenes and/or tumor suppressor genes and specific cancer subtypes is increasingly apparent. Together, these data indicate that “genetic addiction”—regardless of whether the genetic abnormality befalls an oncogene, a classic tumor suppressor gene, or a NOA gene—is a critical paradigm for understanding the relationship between genome disruption and neoplastic transformation. An obvious but nonetheless important corollary to this concept is that genetically engineered mouse models (GEMMs) in which neoplasms develop will become an increasingly important tool for linking specific cancer phenotypes to unique genetic signatures.
Phenotypes Linked to a Given Genetic Addiction May Be Similar Across Species
The goal of comparative pathology, and indeed of most biomedical research, is to extrapolate findings from animal studies to better comprehend human disease. Recent data suggest that “genetic addictions” in human neoplasms may be shared by their murine counterparts. The reverse may be equally true, in which case new GEMMs of neoplasia hold substantial promise for unraveling the molecular intricacies of at least some human cancers.
An important multispecies example of a specific genetic addiction leading to a particular constellation of cancer phenotypes may be found in the spectrum of urinary bladder cancers that develop in humans and mice. The main morphological signatures of neoplasia in this organ are papillary outgrowths in benign lesions versus anaplasia and invasiveness in their malignant counterparts. Up to 90% of benign transitional cell tumors of the urinary bladder in humans have an activating mutation of the HRAS1 oncogene. A transgenic mouse model that expresses an activated Hras1 in the urinary bladder epithelium develops urinary bladder tumors with morphologic features identical to those seen in humans, thereby strengthening the genotype-to-phenotype link. 84 In contrast, invasive transitional cell carcinomas in humans rarely have an HRAS mutation and are addicted, instead, to the sustained loss of both the RB and TRP53 tumor suppressor genes. The genotype-to-phenotype relationship again is confirmed in the mouse using a model in which expression of the simian virus 40 (SV40) T antigen, which blocks both RB and TRP53 specifically in the urothelium, leads to induction of invasive transitional cell tumors instead of benign papillary masses. 137,142
Outside the cancer realm, the intimate connection between RAS expression and differentiation of mucin-producing cells is another example of the specific morphologic manifestation of certain genes. In humans, activation of the RAS pathway is implicated in mucin overproduction by intestinal and respiratory epithelia. 62,67 In mice, mucin-secreting cells are also addicted to RAS, even in organs that do not normally produce mucin (such as the prostate gland and the pancreas). Mice expressing activated RAS specifically in the prostate exhibit extensive goblet cell metaplasia in this organ (Fig. 1 ). 114 Knowledge of this connection in human tumors prompted researchers to investigate the impact of Ras function in the unexpected phenotype of intestinal metaplasia seen in the prostate of Trp63-deficient mice. Once again, the role of genetic addiction in promoting a specific phenotypic evolution proved true: intestinal metaplasia in the prostate of these animals was related to activation of the Ras pathway. 59 In the pancreas, a KRAS mutation is associated with a majority of human pancreatic ductal carcinomas (PDACs) 2 and is sufficient to induce PDAC in the mouse. 86 Interestingly, pancreatic intraductal neoplasia (PanIN), which is the precursor of PDAC, harbors a KRAS mutation and also is characterized by extensive mucinous metaplasia of the ductal epithelium. Accordingly, it is not surprising that other malignant neoplasms that arise from mucin-producing tissues, such as colorectal and lung carcinomas in humans, are addicted to KRAS mutations as well. 31 Following suit, carcinoma of the salivary gland, with its prominent mucinous cell population, can be induced by targeting mutated Kras to the keratin 5 promoter in mice. 103
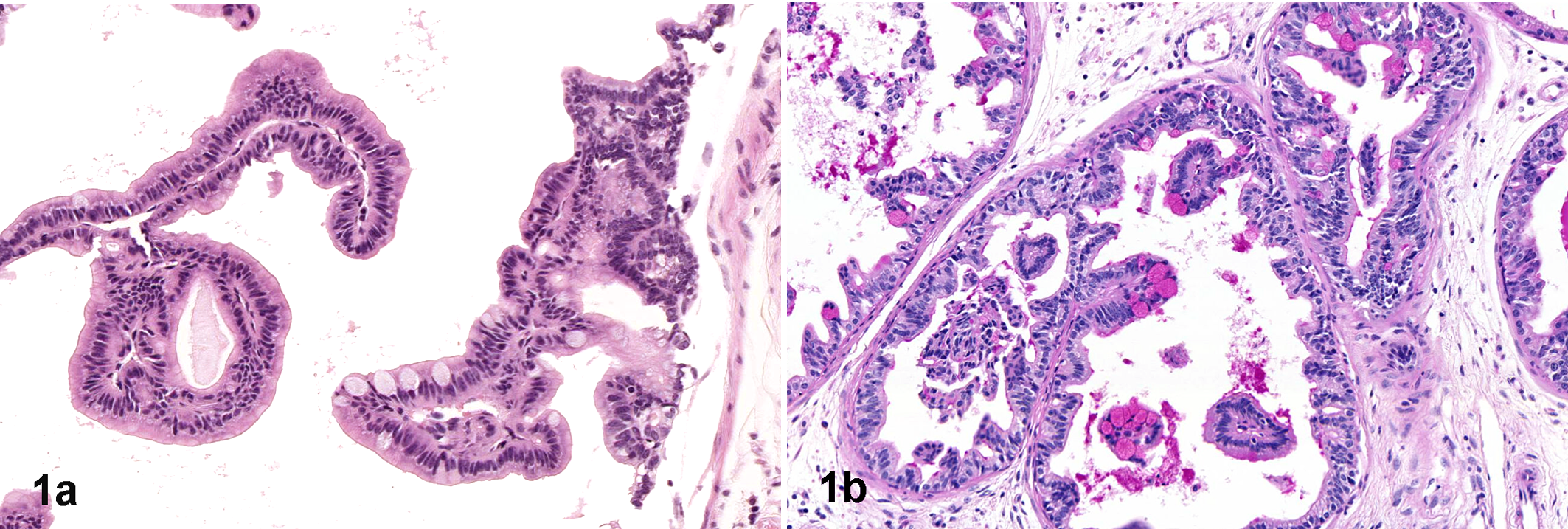
Prostate, 5-month-old probasin-HRas (G12V) mouse (C57BL/6 × FVB background). Ras-associated mucinous metaplasia is indicated by numerous swollen, pale goblet cells (a, hematoxylin and eosin) containing mucin in their cytoplasm (b, pink globules stained for periodic acid-Schiff).
Connections between gene addictions and certain stereotypic phenotypes are more readily identified in GEMMs since the initiating genetic lesion is known. Mouse mammary tumor models provide many good examples of genetic addiction and “pathway pathology.” For instance, tumors generated by the Wnt1 transgene and other genes in the Wnt pathway, such as Wnt10b, Ctnnb1 (β-catenin), and Gsk3β, have heterogeneous but essentially similar morphologic features, including a combination of acinar and glandular patterns, abundant stroma, areas of squamous metaplasia, and well-organized myoepithelium, a feature that is rarely seen in other mammary tumors (Fig. 2 ). 39,110 The so-called Wnt tumors, which exhibit tubular, glandular, and cribriform patterns, are very different from the distinctive solid carcinomas resulting from a Erbb2- (Her-2/neu) activating mutation of the epidermal growth factor receptor (EGFR) gene (Fig. 3 ). 4,85,110 Another example comes from mice lacking E-cadherin and Trp53, which develop mammary tumors with lobular morphology just like their human counterparts. 24 Importantly, lobular mammary carcinomas, as well as the solid, comedo-carcinomas displayed by Erbb2-mutant mice, closely resemble the human tumor they emulate but are rare as spontaneous lesions in the mouse mammary gland. 4,128
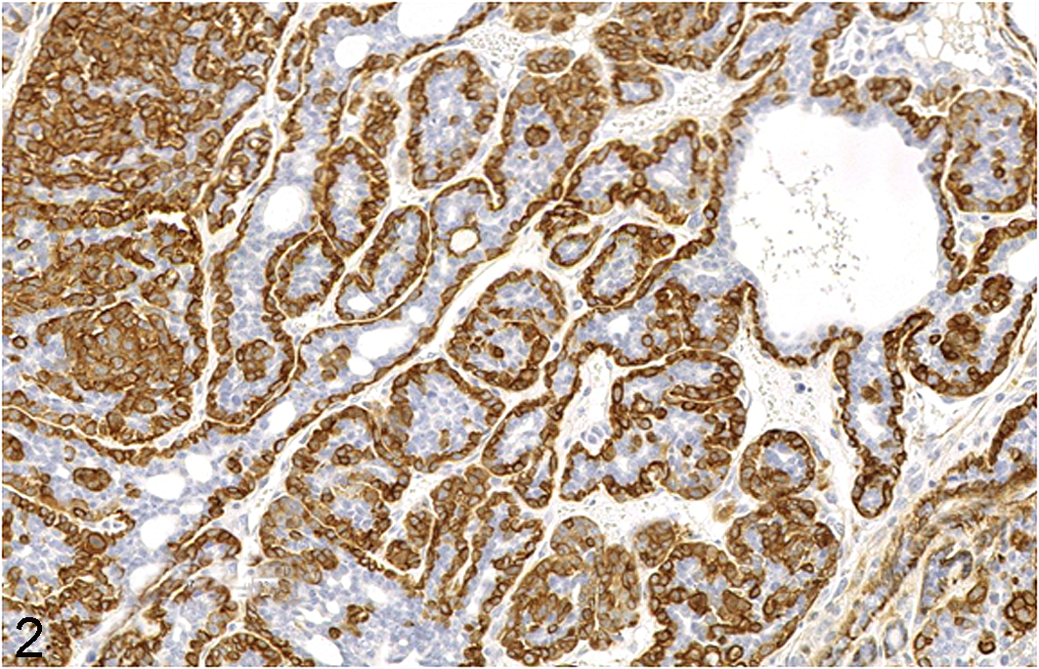
Mammary gland neoplasm, 6-month-old MMTV-rtTA-pA/TetO-Wnt-1/p53–/– mouse (FVB background). This stereotypical Wnt-1 tumor is an adenocarcinoma that exhibits common Wnt-associated traits such as intermingled tubular, glandular, and cribriform patterns with a prominent, continuous myeopithelial layer highlighted by intense α-smooth muscle actin (α-SMA) expression. Continuous α-SMA-positive basal layers are rarely seen in non-Wnt mammary tumors. This pattern of Wnt-related changes is also seen in spontaneous mammary neoplasms induced by mouse mammary tumor virus (MMTV). Anti-α-SMA indirect immunohistochemistry with hematoxylin counterstain.
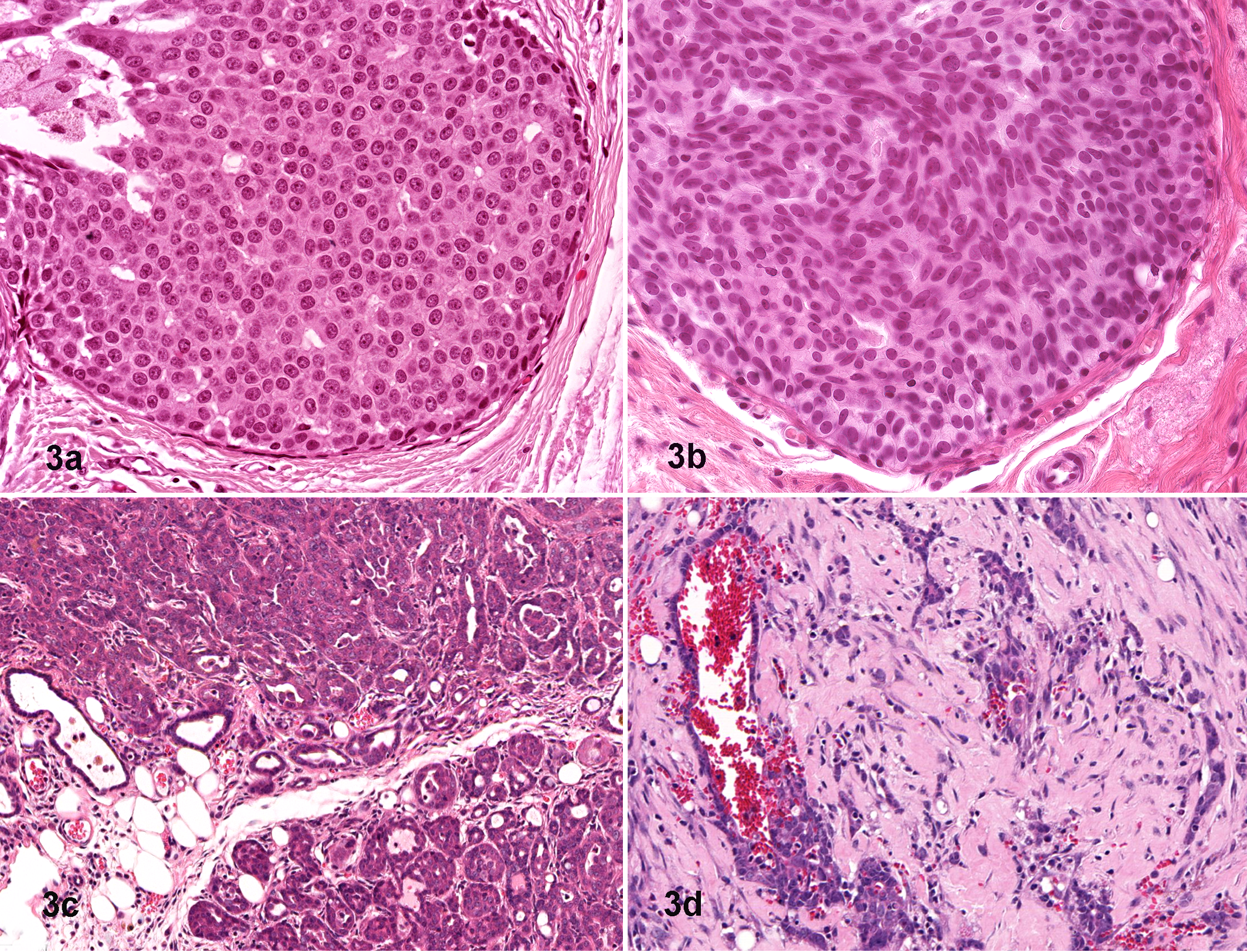
Mammary gland neoplasms, 6-month-old Tg(MMTV-LTR:Neundl) mouse on the FVB background (a) and human (b). Aberrant Her2/neu expression is associated with a solid intraductal growth pattern with no evidence of glandular or tubular differentiation. Similar solid carcinomas develop in tetracycline-controlled Her2/neu conditional transgenic mice (8-month-old, MMTV-rtTA-pA/TetO-Neundl, FVB construct) treated with doxycycline (0.1 mg/ml for 6 weeks) (c) and regress within 48 hours after doxycycline withdrawal (d), clearly demonstrating the principle of oncogene addiction. This tumor type is not seen in mice that spontaneously develop neoplasms following infection with the mouse mammary tumor virus (MMTV). Hematoxylin and eosin.
Sometimes the genetic addiction happens at a broader level and does not necessarily imply a specific morphologic feature as much as an entire tumor phenotype. Burkitt lymphoma, mouse plasmacytoma, and rat plasmacytomas, all of which are driven by a c-Myc habit, 51,143 and human pancreatic adenocarcinoma, with its strong dependence on KRAS mutation, 139 are examples of this broader relationship. Recently, another link between morphologic appearance and genetic constitution has been identified when poorly differentiated (by histopathologic assessment) basal-like breast cancers, glioblastomas, and urinary bladder cancers were found to overexpress 3 genes (Nanog, Pou5f1 [Oct-4], and Sox2) that comprise a genetic signature similar to that of embryonic stem cells. 7 Therefore, a reasonable expectation is that the discerning comparative pathologist will ask whether other poorly differentiated tumors carry a similar molecular signature. Such questions provide the basis for rapid advances in understanding new links between specific genes and particular morphologic patterns.
In summary, an increasing number of genetic addictions will be recognized as data on gene function are collected via phenotyping of new GEMMs. By noting the unique morphologic characteristics of certain gene/cell-type interactions, the pathologist quite often can make rational predictions regarding the possible molecular pathways that might be affected in certain tumor types. Knowledge of these genetic addictions or tendencies may also be useful in interpreting unexpected outcomes when using mouse models to explore mechanisms of human disease.
Tumor Suppressor Gene Addictions
Proteins encoded by tumor suppressor genes have a wide range of functions within the cell, but all have one trait in common: they prevent uncontrolled cell proliferation and growth and therefore decrease the likelihood of cancer development. The 2 prototypical genes in this family are RB, the first tumor suppressor gene to be discovered, 54 and TRP53. The importance of RB is illustrated by the occurrence of RB deficiency or disruption of RB-regulated pathways in many human cancers. 133 In contrast, TRP53 gained its reputation because it lies at the crossroads of many systems designed to detect and repair DNA damage and thus has the ability to halt cell division and eliminate defective cells before they can threaten the health of the organ or the survival of the individual.
Traditionally, tumor suppressor genes have been classified into 2 types, the “gatekeepers” and the “caretakers.” Gatekeepers are the classic tumor suppressor genes, such as RB and TRP53. These genes encode proteins that prevent unruly cell proliferation by halting the cell cycle and/or inducing apoptosis. When compared to RB and TRP53, the more recently identified but extremely important PTEN (phosphatase and tensin homolog) gene has many functions related to cell survival and motility and can also be classified as a gatekeeper because it indirectly activates the apoptotic machinery. In contrast, the caretakers are responsible for maintaining genome integrity and usually function as DNA repair genes; examples include BRCA1 and BRCA2. Some genes with tumor suppressor functions, however, do not fall into either of these 2 categories directly and have triggered the recent recognition of a new tumor suppressor gene class: metastasis suppressors. The prototype of this new division is CD82 (KAI1). 71,120 At a broader level, tumor suppressor genes that may not be sufficient to elicit neoplasia but that play a key role in modifying cancer phenotype can be referred to as NOA genes. 74
The separation of tumor suppressor genes into the above categories has facilitated the understanding of many aspects of cancer biology. However, although the loss of certain tumor suppressor genes can be “seen” under the microscope, the implications of the different categories, if any, remain unknown. In the next section, we explore the “addictive effect” of 3 tumor suppressor genes—RB, TRP53 and PTEN—as seen from the pathologist’s perspective.
RB Addiction Mediates Neuroendocrine Differentiation and Unique Epithelial Phenotypes
Molecular Mechanisms
RB encodes a pocket protein, pRB, that acts as the gatekeeper of an important checkpoint of the cell cycle (the “restriction” or R point) at the end of the G1 growth phase. If the cell progresses past this point, then it is committed to finish the cell cycle, independent of external stimuli. In its active form, hypophosphorylated pRB binds transcription factors of the E2F family. Most of the E2F transcription factors are bound to DNA to the promoter regions of target genes, whose products will then move the cell into the S phase (for DNA synthesis). When associated with pRB, the E2Fs fail to activate transcription of these genes, and the cell either remains in G1 or reverts to the G0 resting phase. Conversely, hyperphosphorylation of pRB renders it inactive and results in the release of the E2F factors and subsequent gene transcription. To regulate this process, the cell counts on the presence of cyclins and cyclin-dependent kinases (CDKs), which act to phosphorylate pRB, and on inhibitors of CDKs, which remove phosphate groups from pRB. 41,99,133
The active form of pRB also represses transcription by remodeling chromatin structure through interaction with proteins involved in nucleosome remodeling, histone acetylation, deacetylation, and methylation. 75 RB gene inactivation is most frequently achieved through chromosomal mutations (such as in retinoblastoma tumor development) followed by loss of heterozygosity 90 that result in complete loss of RB function. Functional inactivation of pRB can also occur by viral oncoprotein binding; this is the main mechanism of pRB loss in uterine cervical cancer, mesothelioma, and Burkitt lymphoma associated with AIDS. 23,35,57 To confound matters further, 2 other pocket proteins from the RB family, named p107 and p130, have similar or overlapping functions to pRB and may play a role in mouse cancer. These 2 RB-related proteins have not been directly implicated in human carcinogenesis. 135
Although alterations of 1 or more proteins within the RB pathway are common in most human cancers, loss of RB itself is mainly associated with sporadic retinoblastomas and small cell lung carcinomas (a neuroendocrine tumor). 36,135 Furthermore, germline mutation of RB results mostly in retinoblastomas and osteosarcomas. This narrow range of malignancies suggests that RB has cell type–specific functions when it comes to tumor suppression. In the mouse, the cell-specific effects of germline Rb mutation are even more remarkable, and a distinctive neuroendocrine preference emerges with manifestations in many organs.
Phenotypic Traits of RB Mutations
Rb knockout mice perish as embryos, but Rb+/– animals have an increased incidence of several neuroendocrine tumors such as adrenal medullary pheochromocytomas, pituitary carcinomas of the anterior and intermediate lobes, and thyroid medullary carcinomas. 45,49,93 Rb-deficient chimeras develop the same tumor spectrum described for heterozygous knockout animals. 136 Neither Rb heterozygous nor Rb–/– chimeric mice develop retinoblastoma. In fact, retinoblastoma fails to develop in mice even when Rb disruption is targeted to the retina via the photoreceptor-specific IRBP (interphotoreceptor retinoid binding protein) promoter. Instead, IRBP-Rb–/– mice develop pituitary tumors and pineal gland tumors. 132 The absence of retinoblastoma in these 3 lines of Rb-deficient mice is likely due to the functional overlap of p107 because the Rb–/–;p107–/– chimera develops retinoblastoma with inner nuclear layer differentiation. 107 In summary, germline Rb deficiency in mice tends to manifest morphologically in the form of neuroendocrine tumors.
Recognizing that, at least in the mouse, neuroendocrine tumors are addicted to Rb deficiency provides ample grounds for asking several important questions. For instance, is Rb involved in the pathogenesis of neuroendocrine tumors in mouse models that are not necessarily based on engineered Rb loss? This possibility has been investigated in Men1+/– mice, which develop the same range of neuroendocrine neoplasias displayed by people who present with hereditary type 1 multiple endocrine neoplasia syndrome because they harbor mutations in the MEN-1 gene. The overlapping phenotype of Rb+ /– and Men1+ /– mice has allowed scientists to identify interactions between these 2 genes in a cell lineage–specific manner. In particular, evidence that MEN1 regulates RB phosphorylation in certain cell types offers clues toward a more concrete link between RB function and neuroendocrine neoplasia. 81
Another important question is whether altered expression of RB family members may lead to the genesis of other tumor types. The premise has been explored using a mouse model of prostate cancer named TgT121. This transgenic line carries a truncated version of the SV40 large T antigen that binds pRB, p107, and p130 simultaneously, but it differs from other SV40 constructs because it does not bind TRP53. 123 The T121 SV40 fragment has been tagged to promoters specific to prostate (ARR2), mammary gland (Wap), and choroid plexus (LPV), at which sites it induces epithelial tumors. 43,73,112 These lesions, although located in 3 very different, nonneuroendocrine organs, share several morphologic similarities, including a tendency toward epithelial dysplasia and a high nuclear-to-cytoplasmic ratio. A connection between Rb and certain features of epithelial proliferation becomes clearer when prostate-specific T121 mice are examined. 44 These mutants exhibit extensive prostatic dysplasia characterized by crowded, tufted epithelium and hyperchromatic nuclei. When these mice are crossed to Trp53–/– mice, a significant degree of stromal proliferation and occasional stromal tumors are added to the repertoire of epithelial lesions. Although T121 mice develop hyperplasias, dysplasias, and microinvasive adenocarcinomas, prostatic conditional Rb knockout animals have minimal lesions, if any, which may be regulated by intact p107 and/or p130 in the latter. 44,76 Interestingly, T121;Trp53 double knockout mice have prostatic lesions identical to those found in TRAMP (transgenic adenocarcinoma of the mouse prostate) mice, which harbor the complete SV40 transgene. 34,115 This finding is not surprising as intact SV40 blocks both pRb and TRP53, which is exactly the genetic background of the double cross mentioned above.
A third important question is to define the nature of the genetic addictions that determine whether animals with Rb deficiencies will develop neuroendocrine tumors, epithelial neoplasms, or both. For example, T121;Trp53–/– mice do not develop neuroendocrine tumors, but such malignancies are common in TRAMP mice. 19 This observation is important because studying the net genetic differences between these 2 models may shed light on the yet unknown molecular pathogenesis of SV40-induced neuroendocrine tumors. 20 Genesis of neuroendocrine neoplasia could be a strain-related phenomenon because T121;Trp53–/– mice have a mixed genetic background with a strong C57BL/6 component; this strain is known to develop neuroendocrine tumors much later and less frequently than FVB mice. 115 Promoter specificity will also need to be considered because the probasin promoter used to target the prostate in the TRAMP model is also expressed in the prostatic stroma, whereas ARR2 (the promoter used in TgT121) is thought to be restricted to the epithelium. 141 Differences in cell lineages suffering the disruption of both tumor suppressor genes also may play a role because Trp53–/– is a germline mutant (lost globally) in the T121;Trp53–/– animals but is inactivated simultaneously with Rb and restricted to the prostate in the TRAMP model.
It is important to keep in mind that identical mutations (i.e. genetic addictions) may exhibit different phenotypic responses in mice and men. Besides the overlapping role of other pocket proteins, RB functions differently in the mouse when compared to humans. Murine cells have many-fold lower levels of RB protein, and the gene does not bind to the promoter regions of many proapoptotic murine genes that are considered its targets in humans. 140 Furthermore, mice with similar genetic addictions but distinct genetic backgrounds may express divergent structural changes. Comparative pathologists and researchers using GEMMs to explore human disease mechanisms should be aware of such differences to correctly translate mouse data to human disease.
TRP53 Addiction Mediates a Spindle Habit
Molecular Mechanisms
In 1979, researchers were using immune sera from mice and hamsters bearing tumors induced by SV40 to study murine cell lines immortalized and transformed by the viral antigens. They found that a protein slightly smaller than 54 kD, subsequently called p53, immunoprecipitated along with SV40 large T and small t antigens. 42 Using the same immune sera, TRP53 was also detected in 2 different, uninfected murine embryonal carcinoma cell lines. Analogous proteins were present in hamster, monkey, and human cells. 69 Subsequently, cDNA cloned from cells with abnormal and wild-type complements of Trp53 (obtained from tumor cells and normal cells, respectively) were introduced, together with the Ras oncogene, into cultured rat embryonic fibroblasts. Although fibroblasts containing a combination of Ras and mutant Trp53 showed large numbers of transformed spindle cell foci, those harboring Ras and wild-type Trp53 exhibited no transformation. These results suggested that the wild-type Trp53 gene was a tumor suppressor gene and that its mutant version was associated with neoplastic transformation. 28
TRP53 responds to many types of injurious stimuli, including DNA damage due to irradiation or genotoxic chemicals, hypoxia, cell starvation or malnutrition (lack of proper nutrients), pH changes, and many other intra- and extracellular stressors by inducing cell cycle arrest and/or apoptosis. 116 Because of its nuclear localization and DNA-binding abilities, the TRP53 protein is considered a transcription factor. It is produced at a constant rate in the cell but has a very short half-life of about 20 minutes. In normal cells, TRP53 levels are very low because the protein is ubiquitinated by MDM2, transported out of the nucleus, and degraded in a proteasome. 83 However, the phosphorylated or activated version of TRP53 becomes stable, cannot be ubiquitinated, and remains in the nucleus, where it can then activate transcription of target genes. For example, ionizing radiation causes double-strand DNA breaks that are sensed by specific proteins that signal through the ATM kinase and its downstream kinase ATR; both of these proteins can phosphorylate TRP53 and protect it from degradation. 47,92 Cell cycle arrest then ensues so that the damaged cell has a chance to repair its chromosomal defect. If the repair is successful, the cell proceeds through the cycle, but if the damage is excessive, the cell is sent down the apoptotic pathway. 102
Most TRP53 functions depend on the activation of target genes. Examples include CKDN1A (P21), a broad-range CDK inhibitor that allows TRP53 to halt the cell cycle in the G1, S, G2, or M phase; GADD45 and XPC, both of which are responsible for nucleotide excision repair to remedy DNA damage; and BAX, BCL2, and PUMA, which regulate apoptosis. Together, these TRP53 targets act by either causing cell cycle arrest or activating the apoptotic cascade. The choice between these 2 fates depends on such factors as cell type, the severity and duration of the stressful stimulus or genetic damage, and the balance between proapoptotic and antiapoptotic factors. Because of its many critical functions and targets, TRP53 is considered the central guardian of the genome. 131
Almost 3 decades after the discovery of the TRP53 protein, mutation of this important tumor suppressor gene has been identified in more than half of all human cancers (as reported in the R15, November 2010 IARC TRP53 database). The IARC TRP53 database carries a compilation of relevant statistics about sporadic and germline mutations, as well as tracks polymorphisms of TRP53. 97 This database also contains information regarding the function and structure of the TRP53 protein, as well as a list of existing cell lines and mouse models used to study this critical tumor suppressor gene. According to this database, the highest prevalence of somatic TRP53 mutations in human neoplasms occurs in solid epithelial tumors affecting organs such as the colon and rectum, esophagus, larynx, liver, lung, ovary, pancreas, and skin. However, germline TRP53 mutations, as in Li Fraumeni syndrome (a rare autosomal dominant, hereditary condition predisposing afflicted individuals to many forms of cancer), present a significantly different tumor pattern.
Phenotypic Traits of TRP53 Mutations
Although breast carcinoma remains the tumor type most commonly associated with germline mutations of TRP53, a high incidence of soft tissue sarcomas and tumors of the adrenal gland and brain are also reported in human patients with Li Fraumeni syndrome, pointing toward a shift from induction of carcinomas to sarcomas, especially those having a prominent spindle cell phenotype. The sarcoma predilection is emulated in the Trp53-deficient mouse. 50
The germline mutation patterns of TRP53 in humans are partially mirrored by Trp53-deficient mice. Mice with homozygous null mutations of Trp53 develop a high incidence of sarcomas, mainly of soft tissues, second in number only to lymphoma. 50 On the other hand, soft tissue sarcomas are the most common malignancy of Trp53-heterozygous mice. 50 Lymphomas in these mice are mainly of thymic origin and are composed of immature, incompletely differentiated T lymphocytes (positive for both CD4+ and CD8+). The different tumor spectra in mice with homozygous and heterozygous deficiencies of Trp53 may be due to the different latencies of neoplasms associated with complete versus partial loss of this tumor suppressor gene because Trp53–/– mice develop tumors much earlier than do Trp53+/– animals. Tumor latency is much longer in heterozygous mice because they require a second inactivating mutation or LOH that affects the remaining wild-type Trp53 allele to develop tumors. Furthermore, the pool of susceptible immature thymocytes prone to neoplastic transformation is much decreased in older animals due to thymic involution, which likely explains the much lower prevalence of lymphomas relative to sarcomas in Trp53+/– mice. 50
Many architectural patterns of sarcomas arise in Trp53+/– and Trp53–/– mice, but most neoplasms have an overall spindle cell morphology. Tumors such as fibrosarcomas, leiomyosarcomas, and rhabdomyosarcomas with their expected fusiform morphology are common in these animals. However, even hemangiosarcomas and osteosarcomas often have a predominantly spindloid appearance in Trp53 mutant mice; hemangiosarcomas can be predominantly solid masses of spindle cells with a “bloody appearance” imparted by the presence of relatively few small, tortuous, and indistinct vascular channels, whereas osteosarcomas often appear to arise as a fibroblastic mass that contains a few small foci of osteoid and/or bone production. These observations support a close link between the loss of Trp53 function and a predilection for spindle cell differentiation in mice.
A remarkable illustration of TRP53’s propensity to mediate a spindle habit in neoplastic cells is its connection to a specific cancer phenotype called epithelial-to-mesenchymal transition (EMT) type tumors. Another designation for these entities often used in diagnostic reports for human tumors is carcinosarcoma. Evidence of a role for Trp53 in the pathogenesis of EMT tumors in the mouse came from several mouse models in which spindle cell masses with unique immunohistochemical features arose in the mammary gland. 15 The neoplastic spindle cells in these unusual tumors often simultaneously coexpress markers specific for both epithelial and mesenchymal differentiation, such as cytokeratins 8/18 in conjunction with smooth muscle actin (SMA) or vimentin (Fig. 4 ). Evaluation of cytokeratin expression in these tumors confirms the existence of an indistinct glandular epithelioid pattern (as seen in sections stained with hematoxylin and eosin) within an otherwise spindle cell–rich mass. Characteristic EMT tumors were frequently found in crosses of various mouse mammary tumor models, such as mice with Brca1– /– or Hus +/– mutations, onto the Trp53 +/– background. 22,72 However, cancers with the EMT phenotype were also noted in GEMMs without original disruptions in Trp53 function, such as animals with systemic null mutations of Gzmm (Met-1) and Met-1AS-OPN or, more recently, mice with targeted deletion of Brca1 or Bard51 in the mammary gland epithelium using an inducible mouse mammary tumor virus (MMTV) promoter (unpublished data). 52 These observations are relevant because EMT tumors are rare in wild-type mice, which further emphasizes the causal relationship between Trp53 mutations and spindle cell differentiation in mouse tumors.
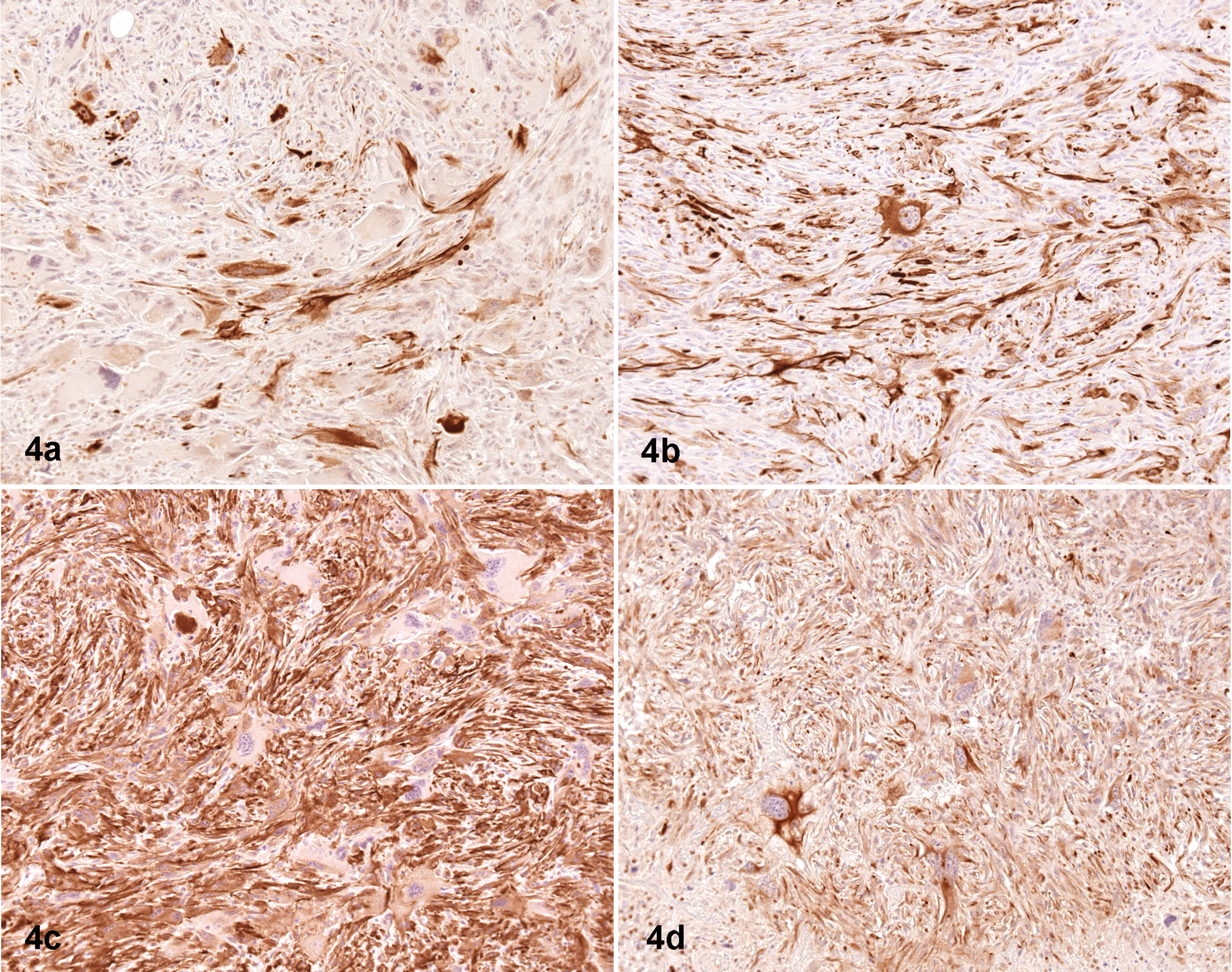
Mammary gland neoplasm, 9-month-old p53+/–;Hus1 +/– mouse (mixed C57BL/6, S129 background). A common phenotype of the aberrant Trp53 genotype is spindle cell differentiation, or the epithelial-to-mesenchymal transition (EMT) phenotype. The distribution of 2 epithelial markers—keratin-14 (a) and keratin-8 (b)—overlaps with the location of 2 mesenchymal differentiation markers—α-smooth muscle actin (c) and vimentin (d). Importantly, the neoplastic epithelioid and spindle cell populations both abundantly express 2 or more of the intermediate filaments and α-smooth muscle actin. Indirect immunoperoxidase procedures with hematoxylin counterstain, performed on serial sections.
As with the link between Rb disruption and the neuroendocrine phenotype, the recognition of TRP53’s spindle habit brings forth additional lines of inquiry. One obvious need is to explain how the link between cell shape and TRP53 abnormalities is regulated from the molecular perspective. In the case of the EMT phenomenon, it is reasonable to suggest a possible connection between Trp53 and genes known to be involved in epithelial-to-mesenchymal transitions, such as the transcription factors Snai2 (Slug), Snai1 (Snail), and Twist1. In support of this reasoning is the fact that TWIST1 has been recently shown to interact directly with TRP53, disturbing its tumor suppressor gene functions. 117 It will also be important to investigate the status of adhesion molecules such as E-cadherin in EMT tumors because Snail blocks E-cadherin expression. 12 Doubtless, a combination of molecular changes that participate in mediating the effects of Trp53 deficiency will be defined during the course of future investigations in this field.
Other epithelial tumors in humans with tendencies toward spindle differentiation, such as adenomyoepitheliomas or mammary gland carcinosarcomas, also harbor TRP53 mutations. 5,40,101 Aggressive myoepithelial carcinomas of the salivary gland with their poorly differentiated spindle cell morphology as well as colonic, cutaneous, esophageal, gastric, ovarian, and uterine carcinosarcomas are also addicted to TRP53 disruption.* Furthermore, TRP53 mutations are found in sarcomatoid transformation of carcinomas of the kidney and prostate, in sarcomatoid differentiation of squamous cell carcinomas of the skin, and in sarcomatoid tumors of the upper respiratory tract and liver. 6,17,26,46,87,94
In summary, comparative pathologists faced with a predominantly spindle cell conformation in neoplasms should be prompted to investigate the integrity of the TRP53 gene. Entities subject to such directed assessment will include sarcomas, carcinomas with spindle cell morphology, and EMT tumors of many organs in both mouse and man. Nuclear features of spindle cell tumors may also provide a clue that TRP53 function has been affected. This latter observation comes from the fact that loss of TRP53 function results in the perpetuation of genomic damage and aneuploidy, which often manifests morphologically as increased nuclear atypia. 43
PTEN Addiction Promotes an Inflated Cell Image
Molecular Mechanisms
A relatively new player, the gatekeeper gene PTEN, has quickly risen to the forefront of cancer research as a critical tumor suppressor gene because of the increasing evidence for its active participation in numerous human cancers. A high frequency of LOH on chromosome 10q (later narrowed to the bands at 10q21-25) has been demonstrated in many human cancers, including endometrial carcinoma, glioblastoma, malignant melanoma, meningioma, prostate adenocarcinoma, renal cell carcinoma, thyroid follicular carcinoma, and transitional cell carcinoma of the urinary bladder. † Almost simultaneously, the gene responsible for Cowden disease, an autosomal dominant inherited syndrome that results in multiple hamartomas and a variety of cancers, was localized to the same chromosomal arm. 68,80,91 Further studies of tumors from patients with Cowden disease led to the discovery of a new tumor suppressor gene at 10q23 by 2 independent groups; the proposed names were PTEN (phosphatase and tensin homolog) and MMAC1 (mutated in multiple advanced cancers 1). 64,66,119 Shortly after its discovery, PTEN mutations also were linked to Bannayan-Riley-Ruvalcaba syndrome and Lhermitte-Duclos disease, 2 other hamartoma disorders in humans related to Cowden disease; PTEN was also confirmed to be mutated or deleted in many human tumors. ‡ Recent investigations in this field have implicated PTEN in the genesis of several other human neoplasms—adrenal medullary pheochromocytomas and ovarian and mammary tumors—as well as 2 other autosomal genetic disorders, Proteus syndrome (the “elephant man” disease) and Proteus-like syndromes. 55,111,129
PTEN is a membrane-bound lipid phosphatase that inhibits the phosphatidylinositol 3-kinase (PI3K) signaling pathway. Upon activation, PI3K converts the precursor phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-triphosphate (PIP3), an active second messenger that promotes cell survival, growth, and proliferation by activating downstream kinases such as AKT and JNK. PTEN functions to remove a phosphate group from PIP3, thereby converting it back to its inactive PIP2 form and blocking PI3K downstream signaling that leads to enhanced cell growth and division. 77 The AKT kinase isoforms boost cell metabolism and promote both cell survival and angiogenesis by phosphorylating numerous important proteins, including MDM2 (a TPR53 antagonist) as well as BAD and CASP9 (both involved in apoptosis), among many others. 78 JNK, also known as mitogen-activated protein kinase 8 (MAPK8), is important in transcription regulation, cell proliferation, and differentiation. 10 PTEN also plays a role in regulating cell polarity and migration, perhaps via interaction with focal adhesion kinase (FAK). 33 More recently, a role of PTEN in the maintenance of stem cell health in some tissues has also been reported. 113
Phenotypic Traits of PTEN Mutations
Although PTEN does not seem to have a preference for a certain tumor type, its deficiency as a causative and/or contributing event in many hamartoma syndromes unveils a gene addiction that manifests morphologically in tissue overgrowth resulting from more and bigger cells. For instance, Lhermitte-Duclos disease, often associated with Cowden disease, consists of partial macrocephaly with nonneoplastic enlargement of the cerebellum and part of the cerebrum, as well as marked hypertrophy of neurons and glial elements in the affected regions of the brain (Suppl. Fig. S1). 108 In addition, cells lacking PTEN exhibit significantly more metabolic activity and therefore become bigger. The enlargement is mostly due to an abundant cytoplasm with a slightly foamy appearance, likely due to glycogen accumulation resulting from activation of glycogen synthase kinase 3 (GSK3), which is regulated by PI3K. 13 In contrast to lesions caused by TRP53 loss, where the diminishment of gene repair mechanisms leads to considerable nuclear atypia, the nuclear features in these “inflated” PTEN-deficient cells are usually unaltered. This bland nuclear appearance is consistent with the proposed mechanism of PTEN, where disruption leads to a cellular “metabolic” dysfunction localized to the cytoplasm rather than accumulation of genetic damage within the nucleus.
Altered PTEN expression results in a spectrum of hamartomatous changes that may have profound effects in the developing organism. Mice lacking Pten die in utero, whereas Pten+/– animals develop numerous hyperplastic and neoplastic lesions involving a number of tissues, including adrenal and thyroid glands, endometrium, the gastrointestinal tract, liver and gallbladder, lymphoid tissues (mainly lymph nodes and thymus), prostate, and the gonads (germ cells and gonadostromal tumors). 25,100,118,122 In the mouse mammary gland, lack of Pten (Pten–/– conditional knockouts) leads to the development of adenomyoepithelioma, another benign lesion. 65 Although these lesions can reach large sizes, they mostly retain a benign or preneoplastic appearance and behavior unless further genetic changes ensue, such as the loss of the remaining Pten allele or combined loss of a Pten allele and another tumor suppressor gene, as demonstrated by compound prostate cancer models such as the double conditional Pten–/–;Trp53– /– mice and Pten+/–;Nkx3-1+/– mice. 1,18 This observation is in line with Pten’s role as a regulator of hyperplasia and hypertrophy. Furthermore, the diverse lesions in Pten+/– mice display characteristic large cells with slightly foamy cytoplasm (Suppl. Fig. S2). In fact, even normal (nonneoplastic) metabolically active cells in Pten+/– mice, such as hepatocytes and renal tubular epithelial cells, often have abundant foamy cytoplasm. Organomegaly does not come as a surprise in Pten+/– mice due to the widespread presence of cytomegaly. 90,121
Further evidence of PTEN’s inflated idea of the cell comes from its role in mediating cardiac muscle hypertrophy, vascular smooth muscle hypertrophy, mesangial hypertrophy in diabetic nephropathies, and hypertrophy of neurons and astrocytes. 105 Here again, consideration of PTEN’s mode of action may help the pathologist understand resulting morphologic manifestations and recognize the possible involvement of this gene or components of its pathway, such as AKT, in the development of certain diseases and cancers.
Final Remarks
The evolution of modern pathology has followed the many technological advances that have developed since the use of the first simple microscopes in the 17th century. The subsequent discovery of the cell as the functional unit of living organisms, as well as detailed descriptions of tissue morphology during health and disease, characterized the Renaissance of the biological and medical sciences throughout the 19th century. Today, the marvels of molecular technology appear, at times, to clash with traditional histologic examination as the primary tools for investigating disease. Glass slides are being slowly replaced by digital images as the main platform for diagnosing morphologic changes in diseased tissue, and the traditional blue and pink of the classic hematoxylin and eosin–stained section are slowly being supplanted by multicolor “in silico” genomic and proteomic arrays. The gene, and no longer the cell, is considered the functional unit of all living organisms.
That said, the recognition of often subtle differences in color, shape, and size made the discipline of pathology a cross between straightforward science and changeable art. Technology, from the first histochemical stains to the multitude of in situ molecular biology tools currently in use, has helped to transform pathology into a more exact science. Without molecular pathology, morphology is limited, but the reverse is also true. Homogenizing tumors to retrieve DNA, RNA, or protein without knowledge of its morphologic features thrusts us back into the Galenic age of mere lumps and bumps, albeit with a genomic spin. Exact quantification of a genomic signature without considering how the genes relate to structural characteristics is of limited value. It remains important to continually recall that morphology is a result of gene function and that morphology and genetics complement each other on the road to a broader and deeper understanding of cancer and many other diseases.
The goal of this review was to bring to light burgeoning new knowledge regarding the connections between gene expression and particular morphologic phenotypes in cancer biology. By bringing some of these correlations to the forefront, we hope to have heightened the awareness and teased the curiosity of the comparative pathologists engaged in genetically engineered mouse phenotyping and translational medicine. Here we focused on the 3 most important tumor suppressor genes that have been identified to date to exemplify the power of understanding gene-specific structural changes. In both mice and humans, these gatekeeper genes induce fairly stereotypical features: RB is related to neuroendocrine differentiation, TRP53 is highly expressed in spindle cell neoplasms, and PTEN is associated with neoplastic and nonneoplastic expansion and enlargement of many cell types. Despite these trends, it is important to remember that there will be many neuroendocrine tumors with a fully functional RB, carcinomas with spindloid features that have intact TRP53 function, and hypertrophic organs and tumors with large foamy cells that have perfectly normal PTEN function. The key is to make the best use of each traditional morphologic and modern molecular tool to better comprehend how genetic changes affect structural traits and how anatomic features can be used to predict the genetic defects that may contribute to disease. It is the comparative pathologist’s responsibility to learn the shades and hues that link aberrant gene function to specific morphologic patterns to contribute effectively to phenotyping the many new genetically engineered mouse strains created every day and to more rapidly affect efforts to reduce and prevent human cancers.
Footnotes
Acknowledgements
We thank Dr. Gerald Cunha (UCSF) for providing the laboratory space in which some of the genetic addictions described in this manuscript were explored. We thank the Armed Forces Institute of Pathology (AFIP) for making the whole-mount histologic section of the Lhermitte-Duclos disease brain (Suppl. Fig. S1) available for public use. The CD-ROM Version of the Atlas of Tumor Pathology) contains US government work that may be used without restriction. A link to the image within the Pathology Educational Instructional Resource (PEIR) digital library can be found here: http://peir2.path.uab.edu/pdl/dbr.cgi?db=images&uid=default&Collection=*&Collection2=*&Type=*&Type2=*&Description=&syn=num&mh=20&hits=20&File=00405804&view_records=View+Records&view_records=Search.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research and/or authorship of this article: A portion of this work (RDC) was supported by the U01 CA141541, U01 CA141582, and U01 CA105490 grants from the National Cancer Institute’s Mouse Models of Human Cancers Consortium.