
Abstract
Malignant soft tissue tumors are commonly observed in wild-type and gene-targeted mice. These tumors have different degrees of differentiation, cellularity, cellular atypia, nuclear pleomorphism, normal and abnormal mitosis, and giant tumor cells with enlarged polylobulated nuclei. They are often diagnosed as pleomorphic sarcoma, undifferentiated sarcoma, fibrosarcoma, malignant fibrous histiocytoma, sarcoma, or sarcoma, not otherwise specified. Pleomorphic sarcomas have no morphological differentiation toward a differentiated mesenchymal or other tumor type in hematoxylin and eosin–stained sections. With the use of immunohistochemistry, human and mouse, tumors associated with these broad nonspecific diagnoses can often be demonstrated to be of a specific cellular lineage. With mouse models being used to delineate the molecular mechanisms, pathogenesis, and cellular origin of human sarcomas, it will be necessary to correlate the morphological and cellular lineage and the molecular profiles of the pleomorphic tumors associated with these mouse models. The results presented here show that with the use of immunohistochemistry, the cellular lineage of many mouse tumors with pleomorphic features can be determined.
Soft tissue sarcomas are observed in wild-type, genetically targeted mice and mice treated with chemical compounds 1,4,18,33,40,47,62,66,72 and often comprise between 20% and 60% of the neoplasms observed in gene-targeted mice. 60 For both veterinary and human pathologists, the diagnosis of sarcoma poses a significant diagnostic challenge, and this is especially true regarding pleomorphic tumors, those sarcomatous tumors with diffuse or focal pleomorphism. Pleomorphic tumors are a heterogeneous collection of tumors with a similar cytological morphological makeup composed of a varying proportion of spindle cells and pleomorphic (in cytoplasmic and nuclear size and shape) cells in a variably collagenized extracellular matrix. These tumors may contain areas with features consistent with pleomorphic leiomyosarcoma, pleomorphic rhabdomyosarcoma (RMS), pleomorphic malignant peripheral nerve sheath tumor, pleomorphic liposarcoma, pleomorphic angiosarcoma, undifferentiated pleomorphic sarcoma (previously called pleomorphic malignant fibrous histiocytoma), pleomorphic carcinoma, and anaplastic lymphoma. Because of diagnostic uncertainties, many of these tumors may be diagnosed as the collective term sarcoma in mice. 16,32
Until recently, undifferentiated pleomorphic sarcoma previously diagnosed as malignant fibrous histiocytoma was the most frequent human soft tissue sarcoma. 22 Many of these tumors are now more adequately categorized as other tumor types with the use of immunohistochemistry. 23 This article illustrates the utility of immunohistochemistry in delineating the cellular lineage of sarcomas, especially pleomorphic sarcomas, in wild-type and genetically engineered mice (GEM).
Material and Methods
Case Material
The veterinary pathology archives of St. Jude Children’s Research Hospital, Memphis, Tennessee, were used for all cases in the report. All the tissues were collected from mice associated with approved studies by the St. Jude’s Institutional Animal Care and Use Committee. The mice included wild-type mice and genetically engineered mice of various lines associated with cell cycle regulator or tumor suppressor genes. This review is based on our experiences with hundreds of these tumors and is not a compilation of tumor incidences or relative immunohistochemical (IHC) findings. The immunohistochemistry procedure below was conducted as a general diagnostic approach for more than 1000 tumors with sarcomatous morphological characteristics on hematoxylin and eosin (HE) staining.
Immunohistochemical Analysis
Tissue specimens submitted to the IHC laboratory have been routinely fixed for 18 to 72 hours in 10% neutral buffered formalin. Five-micrometer-thick sections of formalin-fixed paraffin-embedded tissue were immunolabeled after heat-induced epitope retrieval. The antibodies used for IHC analysis are listed in Table 1 . A panel of commonly evaluated antigens targeted toward the differential diagnosis is outlined in Table 2 . Appropriate negative controls for each of the antibodies were used. Careful attention was given to positive and negative internal and external controls. Every IHC slide was evaluated for appropriate staining of normal tissue for the antigen in question. Internal positive and negative controls on the same slide as the test tumor completely overruled external controls regardless of whether the external controls were placed on the same slide. This was because specimen fixation is the single greatest variable in IHC analysis. The initial panel used to evaluate the tumor consisted of smooth muscle actin (SMA), muscle specific actin (MSA), desmin, and S-100 protein (S100P) antibodies. Tumors with uniform diffuse labeling with SMA and with MSA and/or any labeling with desmin were subsequently labeled with a myogenin antibody. Specimens that did not label with antibody in this initial panel were subsequently stained with antibody to the remaining antigens in Table 2. Negative controls consisting of replacing the primary antibody with the appropriate species-specific isotype were included for each of the antibodies used.
Types and Sources of Immunohistochemistry Antibodies for Mouse Sarcoma Diagnosis
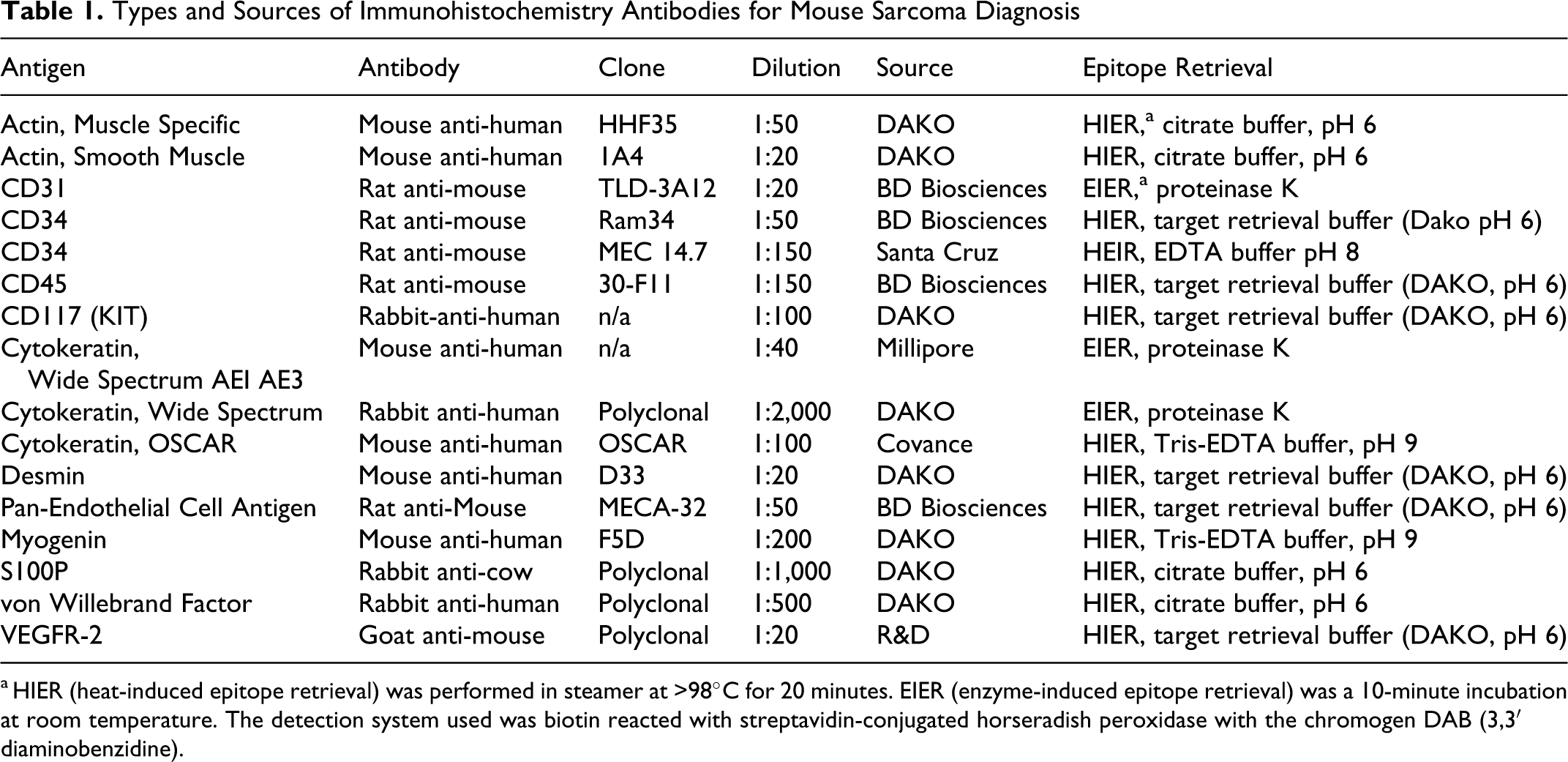
a HIER (heat-induced epitope retrieval) was performed in steamer at >98°C for 20 minutes. EIER (enzyme-induced epitope retrieval) was a 10-minute incubation at room temperature. The detection system used was biotin reacted with streptavidin-conjugated horseradish peroxidase with the chromogen DAB (3,3′ diaminobenzidine).
Immunohistochemistry Tumor Biomarker Profiles for Mouse Sarcoma Diagnosis
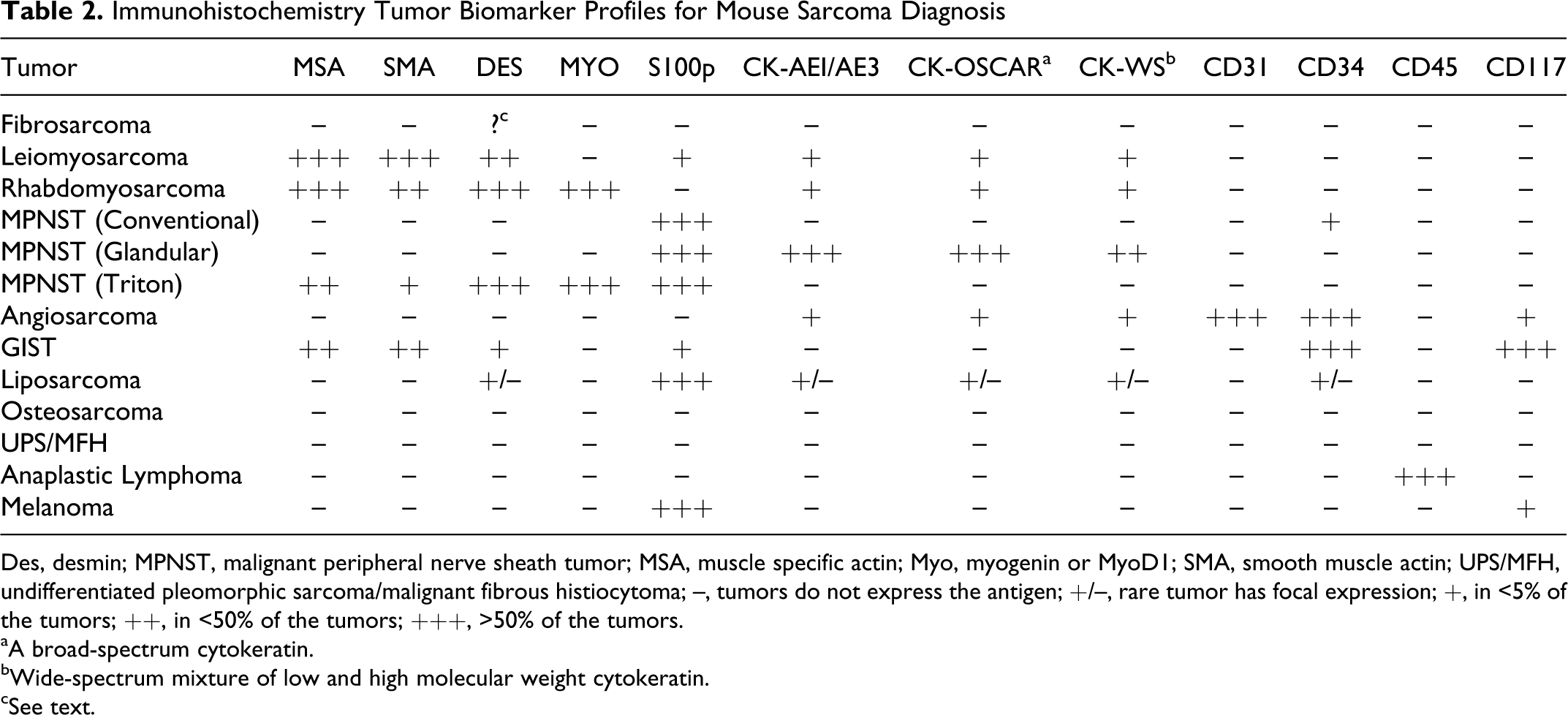
Des, desmin; MPNST, malignant peripheral nerve sheath tumor; MSA, muscle specific actin; Myo, myogenin or MyoD1; SMA, smooth muscle actin; UPS/MFH, undifferentiated pleomorphic sarcoma/malignant fibrous histiocytoma; –, tumors do not express the antigen; +/–, rare tumor has focal expression; +, in <5% of the tumors; ++, in <50% of the tumors; +++, >50% of the tumors.
aA broad-spectrum cytokeratin.
bWide-spectrum mixture of low and high molecular weight cytokeratin.
cSee text.
Results
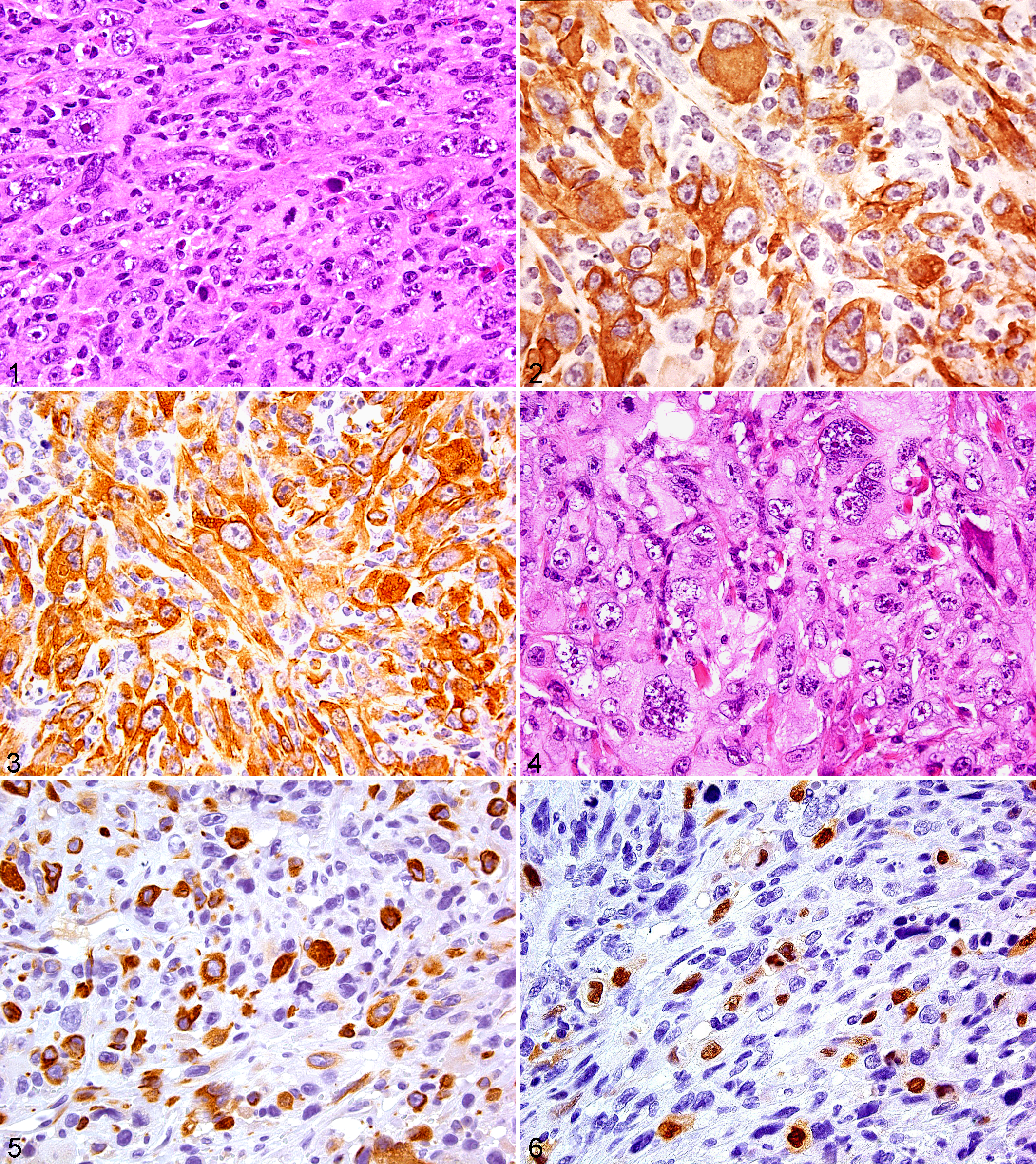
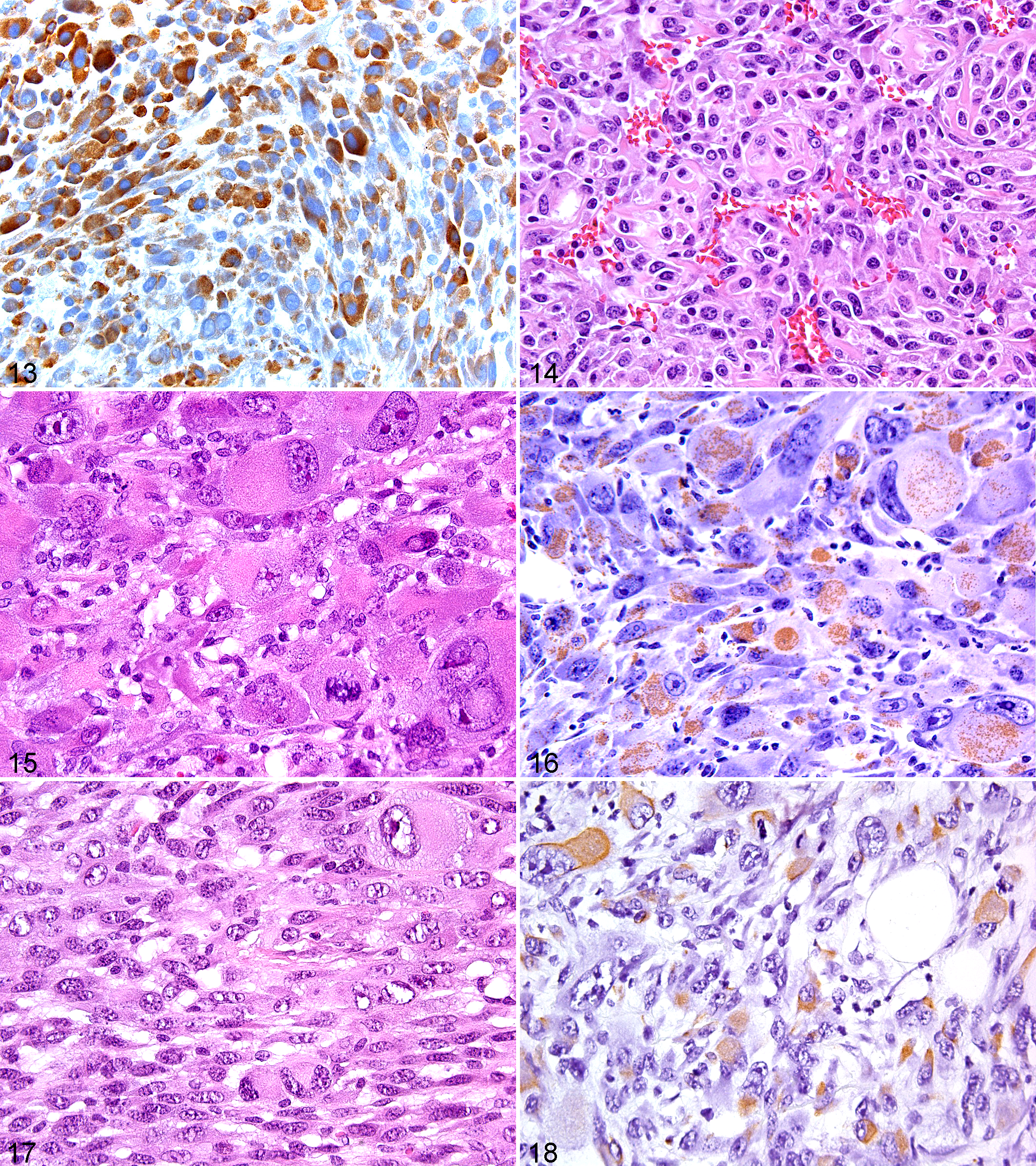
Pleomorphic tumors can occur in various tissues, such as the axial and long bones, skeletal muscle, the dermis, and subcutaneous tissues associated with the chest, back, front and rear legs, and abdominal mesentery. Regardless of cellular lineage, these tumors can consist of similar morphological features consisting of a variable admixture of uniformly spindle and pleomorphic tumor cells set in a sometimes-collagenized extracellular matrix (Figs. 1–18). Several cases (Figs. 1, 4, 10, 15, 17) illustrate the morphological similarity of 5 pleomorphic tumors of different cellular lineages and the obvious diagnostic challenge that HE-stained tissue presents to the pathologist. Based on HE and IHC analysis, these 5 tumors were categorized as leiomyosarcoma, RMS, angiosarcoma, undifferentiated pleomorphic sarcoma, and sarcomatoid carcinoma. In subsequent paragraphs, these tumor categories are more fully discussed.
The monomorphic and pleomorphic mesenchymal tumors include fibrosarcoma, leiomyosarcoma, RMS, malignant peripheral nerve sheath tumor, angiosarcoma, extraskeletal osteosarcoma, liposarcoma, undifferentiated pleomorphic sarcoma (previously designated malignant fibrous histiocytoma), sarcoma, and sarcoma, not otherwise specified (NOS). The nonmesenchymal pleomorphic tumors include spindle cell, undifferentiated or anaplastic carcinoma, gastrointestinal stromal tumor (GIST), and anaplastic lymphoma. In mice, mammary adenocarcinomas 19 and induced prostate adenocarcinomas, which develop into sarcomatous tumors, have been reported. 41,58
Fibrosarcoma
Fibrosarcomas often reveal tumor cells producing collagen, but often spindle cell sarcomas that appear as spindle cells in longitudinal section and round cells in cross section are completely undifferentiated, producing no or little collagen in any locations in the tumor mass. Spontaneous skin sarcomas in mice may have no evidence of collagen production. Sarcomas in GEM often produce no collagen. Differentiating them from undifferentiated sarcomas and other sarcomas (see below) is difficult. Antigens expressed include vimentin and various types of collagen. Masson’s trichrome or Sirius red can show the presence of collagen in the tumor. CD68 has been reported to be expressed in human fibrosarcomas. 69 Whether this occurs in mouse fibrosarcomas has not been shown. However, it has been reported that mouse fibroblasts express some histiocyte markers except for F4/80. 30
Fibrosarcomas do not have any lineage differentiation by IHC analysis. However, there are anecdotal reports that desmin is expressed in some mouse fibrosarcomas. These tumors apparently are not like those of the true human adult fibrosarcoma but are a subpopulation of fibrosarcomas that probably have a scattering of myofibroblast differentiation or a tumor more akin to the human infantile fibrosarcoma. This issue remains to be resolved.
Leiomyosarcoma
Leiomyosarcomas are the most common sarcoma in adult humans. The frequency of leiomyosarcoma in mice is not well documented. They are tumors of smooth muscle in any organ. Soft tissue leiomyosarcomas originate from the arrectores pilorum and the walls of blood vessels normally containing smooth muscle. Leiomyosarcomas in humans and mice have been associated with alterations in genes in the Arf-Mdm2-Trp53 pathway. 33 Poorly differentiated leiomyosarcomas with pleomorphic features are not common and are difficult to differentiate from other mesenchymal malignant tumors (Fig. 1 ). 52
Immunohistochemically, leiomyosarcomas show features of smooth muscle differentiation. Tumor cells have diffuse uniform cytoplasmic expression for α-SMA and muscle-specific actin (Figs. 2, 3). The cells may or may not label with desmin. Although some well-differentiated leiomyosarcomas can be positive for keratin and CD34, pleomorphic leiomyosarcomas do not express these antigens.
Rhabdomyosarcoma
Human RMSs are divided into 3 histological subtypes: embryonic, alveolar, and pleomorphic. The first 2 subtypes are common in children, and the pleomorphic occurs mostly in adults. Rhabdomyosarcomas are a rare tumor in mice with a reported incidence of 14 in 10,000 inbred mice. 66 The chimeric PAX:FKHR genes have been associated with pediatric RMSs, and activation of the 3 RAS isoforms including KRAS has been associated with embryonal and pleomorphic RMS in humans. 65 Similarly, loss of function of the TRP53 tumor suppressor protein has been shown to be a vital event in the progression of human RMS. 51,71 Although RMSs occur in conventional wild-type mice, they are not common, and the molecular pathogenesis of spontaneous RMSs is not known. However, the p16Ink4a/p19Arf-Mdm2-Trp53 and Patched1 pathways have been implicated as a cooperative effect for transgenic and xenotoxic mouse models of all these histological RMS subtypes (Fig. 4 ). 17,20,33,34,59,68,72
Immunohistochemically, RMSs express muscle-specific actin and desmin (Fig. 5 ). They also frequently express SMA. Myoglobin has poor specificity and it is not a good marker for the diagnosis of RMS. Cross-striations are rarely visible in pleomorphic RMS, but well-differentiated tumors in Patched1 (Ptch1) heterozygous mice often have cross-striations. Therefore, a definitive diagnosis of RMS often depends on detection of antigenic activity for either myogenin or MyoD1, which are muscle-specific transcription factors of the MyoD family that are preferentially expressed in the nucleus of differentiating myoblasts. In pleomorphic RMS, myogenin (Fig. 6 ) with nuclear expression and MyoD1 positivity is often limited to a small fraction of the neoplastic cells unlike that in alveolar RMS where myogenin is observed in a high percent of the neoplastic cells. PTAH histochemistry can also help to visualize skeletal muscle striations when they are present in a tumor.
Malignant Peripheral Nerve Sheath Tumor With Pleomorphic Features
Malignant peripheral nerve sheath tumors (MPNSTs) are tumors arising from peripheral nerves or various elements of the nerve sheath (Schwann cell, perineural cell, fibroblast). In human pathology, this noncommittal term replaces the earlier terms malignant schwannoma, neurofibrosarcoma, and neurogenic sarcoma. 69 These terms are still often used for mice. However, in 2004 the Comparative Pathology of Nerve Sheath Tumors in Mouse Models and Humans workshop panel suggested that MPNST tumors in GEM be designated as GEM PNST grade III. 64 Histologically, GEM grade III nerve sheath tumors are similar to the human MPNSTs. The panel chose to use the designation GEM PNST grade III instead of MPNST because the clinical pathological correlations of the mouse tumors are unknown. Although the review authors acknowledge the rationale of the panel for this designation, we prefer to use the term MPNST because it, unlike GEM PNST grade III, denotes distinct morphological features known to the general pathologists.
Loss of the NFI gene has been associated with 20%–50% of sporadic and NFI-associated MPNSTs in patients. 39,44 Inactivation of genes associated with the P16INK4a/P14ARF-MDM2-TRP53 pathways have also been implicated with human MPNSTs. 6,44 Similarly, MPNSTs have been observed to occur in mice with alterations in the p19Arf-Mdm2-Trp53 and other pathways. 9,25,28,70
MPNSTs can arise de novo from a peripheral nerve or progress from a low-grade PNST in the deep fascia or in the area of the spinal or cranial nerves. In mice, this process is commonly seen microscopically in association with a nerve (Fig. 7 ). The tumors may be multinodular or consist of a single nodule. As opposed to human PNSTs, mouse PNSTs are often not encapsulated and often infiltrative. Histologically, MPNSTs display high cellularity, anaplasia, and mitotic figures and may have several growth patterns such as solid diffuse sheets, storiform formations, short and long fascicles that are monophasic, or biphasic fascicles with alternating hypercellularity and hypocellularity. The tumor cells are spindled with hyperchromatic, curved, and tapered nuclei or elongated, blunt-ended nuclei with vesicular chromatin. As in the human tumors, some mouse MPNSTs may display epithelioid, rhabdoid, plasmablastoid, or pleomorphic morphological characteristics and divergent differentiation (bone, cartilage, epithelium, or skeletal muscle). Because of the large spectrum of histological patterns displayed, the distinction of MPNSTs from other malignant tumors of soft tissues may be difficult. For example, the Triton tumor, a MPNST variant with heterologous striated muscle differentiation, can also be confused with pleomorphic RMS and other pleomorphic tumors.
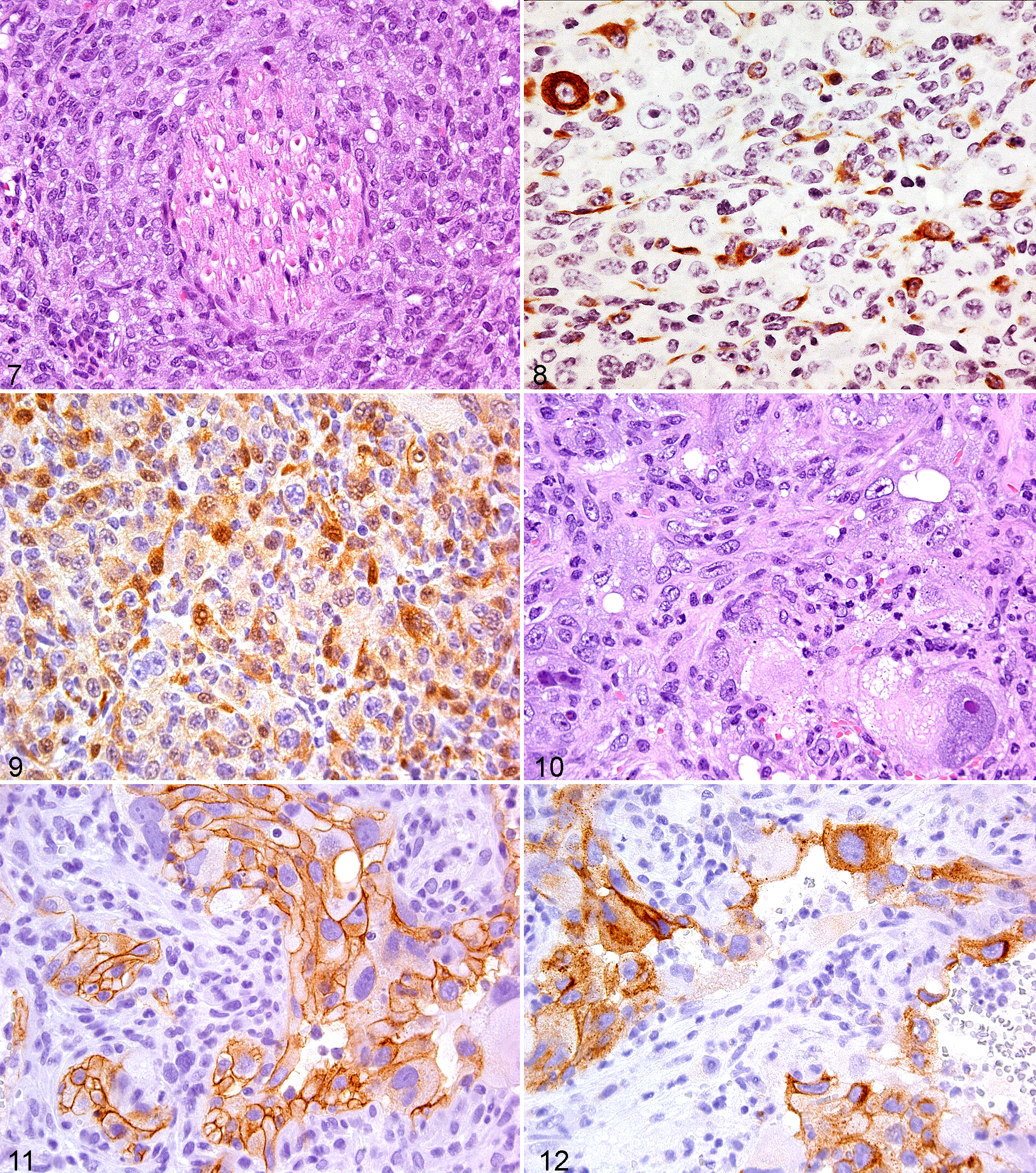
As with the classic conventional MPNST, the epithelioid MPNST will arise in the deep fascia tissue. However, unlike the conventional MPNST, the epithelioid variant also develops in the dermis or subcutaneous tissue. 37 The epithelioid MPNSTs consist of cells with either epithelial-like features or a mixture of epithelioid and spindle cells. The epithelioid cells have round to oval vesicular nuclei with prominent nucleoli. The spindle cells are short and elongated with either hyperchromatic or plump vesicular nuclei. In addition to epithelial-like cells, some epithelioid MPNSTs will have foci of rhabdoid-like or plasmablastoid cells. Cells with pleomorphic features are more common to the epithelioid MPNST than the conventional MPNST.
Conventional MPNSTs have sporadic immunoreactivity for S100P but are negative for SMA and MSA, which are expressed in leiomyosarcomas. Epithelioid MPNSTS have strong diffuse immunoreactivity for S100P, a pattern that contrasts with conventional MPNST. MPNSTs do not express either melanocyte or epithelial markers. A scattering of SMA or MSA positive cells may be seen in MPNST. Occasionally a MPNST with perineurial differentiation will have CD34 expression and epithelial membrane antigen (EMA) immunoreactivity. Unfortunately, in our experience and that of others, EMA immunohistochemistry has been problematic in mouse tissues. 64 In addition, immunoreactivity for desmin (Fig. 8 ), S100P (Fig. 9 ), and myogenin is seen in the MPNST Triton variant with heterologous striated muscle differentiation.
Angiosarcoma (Hemangiosarcoma)
Angiosarcomas generally have defined vasculature differentiation. These tumors have been reported in inbred mouse strains, and in genotoxic and gene-targeted mouse models associated with Kras and mutations in genes of the p19Arf-Mdm2-Trp53 pathway. 1,4,27,60,73 Pleomorphic angiosarcomas consist of a mixture of spindle and pleomorphic cells with poorly defined histological vascular structures (Fig. 10 ). Consequently, it is often not possible histologically to differentiate pleomorphic angiosarcoma from the other pleomorphic tumors discussed in this report unless focal areas of capillary or larger vessel formations are noted. With extensive sampling and diligent histological examination, small foci of vascular differentiation and hemorrhage will often be identified in even the less differentiated angiosarcoma.
Immunohistochemically, these tumors express vascular endothelial markers to various degrees. Therefore, it is necessary to use a panel of antibodies to the endothelial antigens, von Willebrand factor (factor VIII), CD31, CD34, MECA-32 and VEGFR-2 (Figs. 11, 12). Factor VIII is a highly specific endothelial marker. It is observed as fine granules in the endothelial cells, and the endothelial cells secrete it into the circulation. Because of its presence in the circulation, factor VIII can result in high background staining, and it often lines the surface of nonendothelial tumor cells. Consequently, in hemorrhagic areas of a nonvascular tumor, a false-positive interpretation can occur. In addition, platelets express this antigen, and tumor vascular spaces may contain many platelets. Therefore, only tumor cells with intracytoplasmic granules should be considered immunoreactive for factor VIII. CD31 and MECA-32 are more specific than CD34, but MECA-32 antigen has a distribution that is restricted to only small arterioles and venules in the skeletal and cardiac muscle of the adult mouse. 26 Consequently, pleomorphic angiosarcomas arising from skeletal muscle vasculature may not label with MECA-32 antibody. CD31 specificity is excellent, but there are reports of CD31 positivity in human carcinomas and monocytes, which emphasizes the importance for using a panel of antibodies to aid in avoiding a misdiagnosis. 13,43
Gastrointestinal Stromal Tumor
GISTs are mesenchymal neoplasms that arise in the gastrointestinal tract as well as in abdominal soft tissues with no connection to the tubular gastrointestinal tract. Mutually exclusive mutations in CD117, also known as KIT (c-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog), or PDGFR4 (platelet-derived growth factor receptor) are observed in more than 80% of human GISTs. Mutations cause functional changes in CD117 (KIT) and PDGFRA proteins, usually leading to ligand-independent dimerization and constitutional activation. 10,36 Transgenic mouse models with induced constitutional Kit-activating mutations cause Cajal cell proliferation and ultimately GIST mostly in the cecum. 49,56,63
GISTs may have 1 of 4 histological features. Histologically, the tumors may exhibit spindle cell, epithelioid cell, a mixture of both spindle and epithelioid cells, or pleomorphic cell cytomorphological characteristics. Spindle cell GISTs are usually arranged in fascicles, whereas epithelioid cells may be arranged in nests or sheets. The stroma can be hyalinized or myxoid, and blood vessels can be very prominent. The nonpleomorphic GISTs are very monomorphic, with round to elongated nuclei with fine chromatin and inconspicuous nucleoli and abundant cytoplasm.
The key immunohistochemical feature of the GIST is immunoreaction for the c-kit receptor tyrosine kinase, CD117 (Fig. 13 ). Other antigens that are commonly expressed but less GIST-specific are CD34 and nestin. GISTs are variably positive for SMA and S100P, but they generally lack desmin expression. CD117 (KIT) immunoreactivity in GISTs is typically diffuse and strong with a cytoplasmic, membranous, or paranuclear dot-like localization, but expression may be weak and patchy or mosaic with adjacent positive and negative areas. Other abdominal tumors that are variably CD117 immunoreactive are angiosarcomas and melanomas. 45 GISTS do not express endothelial antigens. Melanomas, unlike GISTs, have strong diffuse S100P expression. Keratin immunoreactivity can be seen in GISTs with antibodies reacting with keratin 8 and keratin 18.
Histologically, GISTs can be confused with true smooth muscle tumors, MPNST and UPS-MFH. However, these tumors are negative for CD117 (KIT) immunoreactivity.
Liposarcoma
Liposarcoma is a malignancy of adipocytes. Microscopically, liposarcomas are composed of a relatively mature adipocytic proliferation in which there is significant variation in cell size with nuclear atypia. Varying numbers of monovacuolated and multivacuolated lipoblasts are present, and HE morphological study is the most helpful in the diagnosis of a liposarcoma. Human liposarcomas consist of 5 morphological variants: well-differentiated, myxoid, spindle, dedifferentiated, and pleomorphic. Well-differentiated liposarcomas are reported in mice. 27 Pleomorphic liposarcomas (PLPS) have the histological features common to all pleomorphic sarcomas. As with other pleomorphic sarcomas, alterations (deletions, in activators, mutations, implications, etc) of genes in the Arf-Mdm2-Trp53 pathway have been associated with human PLPS and mouse models of liposarcoma. 42,57,61
PLPS will often have areas positive for S100P, focal stains for SMA, desmin, CD34, and keratins. As previously shown, these immunoreactions are commonly seen in other pleomorphic tumors. Consequently, immunohistochemistry is little help in diagnosing PLPS, and a diagnosis of PLPS relies on the histological detection of malignant lipoblasts with scalloped hyperchromatic nuclei and any foci of fat differentiation. Artifact cytoplasmic vacuolization resulting from fixation may mimic true lipoblasts, leading to a misdiagnosis of PLPS. Consequently, extensive sampling of a tumor is often required to assay for the presence of tumor lipoblasts, and the failure to identify a lipoblast will result in a diagnosis of one or another of the other pleomorphic sarcomas most often UPS/MFH.
Osteosarcoma
Most osteosarcomas of bone origin in genetically engineered mice (Trp53 heterozygotes and others) have foci of early osteoid or late osteoid formation and foci of mineralization of osteoid and are attached to bone. Thus, they can be readily diagnosed. Many, however, can be quite undifferentiated, composed of spindle-shaped or round tumor cells, and tissue sections may be away from the attachment to bone, leading to a diagnosis of sarcoma or undifferentiated tumor of uncertain origin.
Extraskeletal osteosarcoma is a malignant soft tissue tumor showing osteoid formation. Histologically, extraskeletal osteosarcoma has the morphological features typical of a pleomorphic sarcoma except for the presence of osteoid matrix (Fig. 14 ). Many of these tumors have been reported previously as either MPNST with heterologous boney elements or MFH with bone formation.
As reported with other pleomorphic sarcomas in humans, TRP53, RB, and many other genes involved in cell cycle regulation (eg, p14 ARF, p16 INK4a, MDM2) have been consistently altered in osteosarcomas and extraskeletal osteosarcoma. 57 Although extraskeletal osteosarcoma has not been reported in mice, osteosarcomas have been reported to occur in mouse models having loss of tumor suppressant genes such as Arf, Mdm2, and Trp53. 32
Immunohistochemistry for various antigens for the diagnosis of osteoid has not been universally accepted. Presently, the sole acceptable criterion for either osteosarcoma or extraskeletal osteosarcoma is the detection of osteoid production associated with tumor cells as seen in HE-stained sections.
Undifferentiated Pleomorphic Sarcoma (UPS)
Undifferentiated pleomorphic sarcoma is now the preferred term used in human pathology to designate high-grade soft tissue sarcomas that lack any histopathological or IHC evidence of a specific line of mesenchymal differentiation and in which epithelial, melanotic, and lymphoid differentiation has been excluded. 14,21,22 Therefore, UPS is a diagnosis of elimination. Previously, most of these human tumors were called storiform-pleomorphic malignant fibrous histiocytoma.
These neoplasms account for about 5%–7% of sarcomas occurring in adult patients. Tumors with similar cellular morphological characteristics to human MFH have been reported in conventional mice and genetic-targeted mice (Fig. 15 ). 35,46,67 In mice, they are not the same tumors as the common histiocytic sarcomas, although they may on occasion be confused with them histologically. Several investigators have shown with molecular techniques and IHC analysis that many human tumors originally diagnosed as MFH are tumors of other lineages such as leiomyosarcoma, malignant peripheral nerve sheath tumor, spindle cell liposarcoma, malignant melanoma, and sarcomatoid carcinoma. 23,24,50 These tumors were originally thought to arise from facultative fibroblast/histiocytes. It is now recognized these tumors do not arise from histiocytes. 2 The precise molecular or cellular origin of these tumors is unknown. However, these tumors have been frequently associated with inactivation of the RB pathway and alterations (deletions, mutations, amplification, etc) of genes within the P14ARF-MDM2-TRP53 pathway. 5,8
The genetic complexity that characterizes UPS has precluded the development of molecular diagnostics, and histopathological characterization with complementary immunohistochemical staining remains the standard diagnostic method for UPS. Immunohistochemically, UPSs stain for vimentin (Fig. 16 ), but they do not stain for desmin, SMA, MSA, myogenin, S100P, keratins, CD45, or any histiocytic antigens.
Carcinomas With Undifferentiated Pleomorphic Features
Nonmesenchymal tumors with pleomorphic features include carcinomas (Fig. 17 ) and anaplastic large cell lymphoma. Mouse and human carcinomas of the skin and adnexal glands, mammary glands and salivary glands, prostate, and lung have been diagnosed as undifferentiated carcinoma, carcinosarcoma, anaplastic carcinoma, or sarcomatoid carcinoma. They have pleomorphic features, which based on morphological criteria cannot be differentiated from mesenchymal pleomorphic sarcomas with an HE stain, requiring IHC analysis for a definitive diagnosis (Fig. 18 ). Some tumors are histologically biphasic with obvious carcinomatous and sarcomatous areas that contain spindle cell differentiation and large pleomorphic cells with copious amphophilic to eosinophilic cytoplasm and large nuclei and may include a mixture of bizarre large giant tumor cells and eosinophilic cells with rhabdoid features admixed with a spindle fibrous cell matrix. Some of these tumors may show no obvious epithelial areas despite generous sampling. These latter sarcomatoid tumors that have pleomorphic features are indistinguishable from true pleomorphic sarcomas on cytological morphological criteria alone, and IHC analysis is required to delineate epithelial differentiation and a definitive diagnosis. For these possible epithelial to mesenchymal transition (EMT) tumors in mice, mammary 3 and prostate tumors have been characterized. 41,58 For mouse prostate tumors, cytokeratins may be expressed at low levels by sarcomatous tumor cells. 38,41
Myoepithelial carcinomas are tumors of the myoepithelial cell. Because of the plasticity of this cell, myoepithelial tumors have a broad morphological spectrum. These tumors occur in a variety of conventional inbred mouse strains but are most common in BALB/c and A strains. 66 They can also be chemically and transgenically induced in mice. 54,55 Myoepithelial tumors occur in the soft or parenchymal tissues and arise from myoepithelial cells of various exocrine glands (eg, salivary, mammary, clitoral, preputial, and Harderian glands). Unlike human tissue, mouse mammary gland tissue is widely distributed in 10 anatomical locations throughout the body. Consequently, myoepithelial tumors can arise at multiple locations in the mouse, even adjacent to the salivary gland and the tail. In addition to dermal tissue and mammary gland, human myoepithelial tumors have recently been reported to occur in deep intramuscular soft tissues. 29 Acini and duct formations are characteristically absent in a variant of myoepithelial tumors in mice and humans. The malignant myoepithelial tumor of this variant is typically composed of epithelioid and/or spindled cells with cellular pleomorphism consisting of large vesicular nuclei with prominent or large nucleoli, abundant or sparse eosinophilic or lightly basophilic cytoplasm, and somewhat myoid cytomorphological characteristics, rendering these myoepithelial tumors difficult to differentiate from the other pleomorphic tumors. 29,48 Consequently, differentiating myoepithelial tumors from other pleomorphic sarcomas requires immunohistochemistry.
A diagnosis of a myoepithelial tumor requires immunoreactivity for a cytokeratin usually present in the normal myoethelial cells in the tissue of origin on the same slide and at least 1 marker of myoepithelial differentiation. Common epithelial markers used are AE1/AE3, OSCAR, wide-spectrum cytokeratin, and CD138, and markers of myoepithelial differentiation are S-100 protein, SMA, vimentin, and cytokeratins 5 and 14. Myoepithelial tumors will vary in their expression of these markers. Therefore, a panel of 3–5 of these markers is recommended when assessing pleomorphic tumors suspected of being a myoepithelial tumor.
Sarcomatoid carcinomas of the epidermis and adnexa with sarcomatous and pleomorphic features often coexpress cytokeratins, vimentin, and actin. Sarcomatoid squamous cell carcinomas may express almost exclusively high-molecular-weight cytokeratins such as CD5 and CD14. Although the epithelial antibodies AE1/AE3, OSCAR, and wide-spectrum cytokeratin should react with basal keratinocytes (CK5 and CK14), these cells are frequently negative with these antibodies for unknown reasons. Consequently, because of the varied and unpredictable cytokeratin immunoreactivity in these sarcomatoid carcinomas, multiple epithelial markers such as CD138, HMW CK5/6, CK7, and CK14 in addition to the pancytokeratins AE1/AE3, OSCAR, and wide-spectrum cytokeratin may be necessary to demonstrate epithelial differentiation.
Anaplastic Large Cell Lymphoma
Anaplastic large cell lymphoma is a high-grade lymphoma, and a variant of this lymphoma consists of a malignant proliferation of large and often bizarre lymphoid cells. Morphologically, the cells have generous amounts of basophilic or eosinophilic cytoplasm and a variable proportion of nuclei that are horseshoe, kidney-shaped, or multilobulated with prominent nucleoli, and they may have sarcomatoid spindle cell features. When these tumors occur in the cutaneous tissue, they can be easily misclassified as pleomorphic sarcoma. 7 , 15 , 31 , 53 These tumors are rare in mice.
When the differential diagnosis of a pleomorphic tumor in a mouse has been narrowed to either a UPS or an anaplastic large cell lymphoma, the tumor should be stained with a panel of antibodies to lymphoid markers. CD45 (leukocyte common antigen) is known to have high sensitivity and specificity for lymphoid tumors. However, exceptions have been documented. Therefore, it is recommended that in addition to including CD45, the panel should include CD3, CD43, PAX5, IRF4, and vimentin.
Melanoma
Melanomas are rare in wild-type mice but can be readily induced in a variety of GEM. 11 Melanomas can be pigmented or nonpigmented, and high rates of metastasis to lymph nodes or lung have been reported in some models. Nonpigmented tumors can be composed of spindle cells, round cells, and pleomorphic cell populations. If the inducing gene manipulation protocol is known, then the induced tumors, although nonpigmented, and undifferentiated or pleomorphic, are typical of the histomorphological characteristics of the melanomas reported. They often express S100P and tyrosinase proteins to various degrees. 12
Summary and Conclusions
Sarcomas with pleomorphic morphological characteristics that occur in conventional and gene targeted mice comprise a heterogeneous group of tumors, which with the use of immunohistochemistry can be diagnosed as to their cellular lineage except for undifferentiated pleomorphic sarcoma, which is a diagnosis of exclusion. This review discusses and illustrates the morphological and immunohistochemical profiles of mouse sarcomatous tumors of various cellular lineages. Select examples of these tumor types were used to illustrate how immunohistochemistry can aid in determining the cellular lineage of tumors with even the most pleomorphic features. As more mouse soft tissue tumors are subjected to IHC analysis and their molecular pathogenesis becomes more understood, further modification to the classification of these tumors will be warranted, as has been the case with human soft tissue tumors over the past 10 years. The use of the terms sarcoma, NOS or pleomorphic sarcoma, NOS will continue to be warranted when poorly differentiated spindle cell tumors are poorly fixed, when an immunohistochemical analysis is not performed, or when IHC analysis fails to elucidate a cellular lineage consistent with the cytomorphological status of the tumor.
Footnotes
Acknowledgements
We thank Dorothy Bush for her excellent immunohistochemical services, which were invaluable.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This publication was supported in part by grant P30C CA 21765 from the National Cancer Institute and by the American Lebanese Syrian Associated Charities (ALSAC). Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the National Cancer Institute.