
Abstract
Macroautophagy is a cellular degradation mechanism that involves the delivery of cytosolic components (macromolecules or organelles) by the autophagosome to the lysosome for degradation. In mammalian cells, macroautophagy and the ubiquitin proteasome system are 2 major mechanisms to eliminate abnormal proteins accumulated in pathological conditions. Here, the coordination of the 2 pathways to alleviate endoplasmic reticulum stress is reviewed. Also discussed is the regulatory role of macroautophagy and proteasome activity in cell survival and death, as well as the recent discoveries leading to novel strategies of simultaneous control of the proteasome and autophagy activity in anticancer treatment.
The 2 main cellular degradation systems in eukaryotic cells are the ubiquitin–proteasome system (UPS) and the autophagy–lysosome system. Misfolded proteins can be degraded via either pathway. Proteins may be grouped into 3 types according to their functional status and degradation pathways. 20 Type I refers to fully functional proteins that have short half-lives and are degraded at a given step of a normal physiologic process, such as cell division, gene transcription, signal transduction, and endocytosis; here, protein degradation is part of a regulator mechanism. Type II corresponds to nonfunctional misfolded proteins that are degraded as a part of the clearance process. Type III also includes the functional cytoplasmic proteins, which are long-lived, however. The degradation of these proteins is more in response to a changing cellular environment, such as reduction of nutrients. From the functional viewpoint, the UPS can degrade type I and II proteins. Autophagy is mainly responsible for the degradation of type II and III proteins. Just as the degradation of type I proteins can often reflect the functional status of the proteasome, the degradation of long-lived proteins (type III) has been used as a parameter in assessment of autophagic activity.
The UPS system features 3 types of enzymes: ubiquitin (Ub) activating enzyme (E1), Ub conjugating enzyme (E2), and Ub ligase (E3). E1 activates Ub molecules by ATP-dependent adenylation and transfers the adenylated Ub to E2. E3 recognizes specific misfolded proteins or proteins to be degraded and facilitates the transfer of Ub from E2 to the target proteins. There are hundreds of E3 Ub ligases in mammalian cells; each reacts with specific types of substrates, underlying the selectivity and specificity of the UPS system. Continued processing by the Ub-conjugating system generates polyubiquitinated proteins. The ubiquitinated proteins are recruited to the 26S proteasome complex, a multicatalytic enzyme complex composed of the catalytic 20S core and the 19S regulator. In mammalian cells, the 20S core complex has 3 catalytic subunits, which have 3 distinct peptidase activities: chymotrypsin-like, trypsin-like, and caspase-like activities. 31 Currently available proteasome inhibitors interfere with the chymotryptic-like activity of the 20S proteasome. Polyubiquitinated proteins are degraded to peptides by the proteasome, and the freed Ub is recycled. A few proteins are not degraded but transformed from the precursor form to the activated form via the UPS pathway. One such example is the nuclear factor κB (NF-κB), which is freed from the inhibitory protein κB (I-κB) after proteasome processing and is located to the nuclei hereafter. 41 UPS is a mechanism conserved from archaebacteria to humans, and the Ub is one the most slowly evolving proteins, suggesting the pivotal role of the Ub-mediated pathway in basic cellular functions. Many Ub-like proteins have been discovered; however, the physiologic significance of many of these proteins is still unknown.
The autophagy–lysosome system comprises three types—macroautophagy, microautophagy, and chaperone-mediated autophagy—based on the difference in the substrate-to-lysosome delivering mechanisms. This review focuses on the role of macroautophagy (hereafter referred to as autophagy) in the regulation of misfolded protein degradation. In this process, membranes from undefined sources enclose part of the cytoplasm containing macromolecules and/or organelles—including mitochondria, ribosomes, peroxisomes, and endoplasmic reticulum (ER)—to form autophagosomes, also called autophagic vesicles (AVs), which fuse with the lysosomes for degradation. 98
The biogenesis of the autophagosome can be divided into 3 phases: initiation, elongation, and maturation. There are 32 autophagy-related (Atg) proteins defined in the yeast, many of which have mammalian homologues. 46,70 These molecules participate in different phases of autophagosome biogenesis. The initiation step of autophagy is structurally defined as the generation of the crescent-shaped membranous structure called phagophore, or isolation membrane (IM). Because many different stimuli may induce autophagy, there are potentially different signaling pathways involved. 81,86,89 In mammalian cells, the main upstream controlling switch for autophagy initiation is the mammalian targets of rapamycin (mTOR). Two downstream protein kinase complexes are known to be centrally involved—the Atg1/Unc-51-like kinase 1/2 (UKC) complex and the Atg6/Beclin1–class III phosphatidylinositol-3-kinase (PI3K) complex. The UKC includes Atg13 and FIP200 and is activated by the suppression of mTOR, which may lead to the formation of early autophagosome membrane (IM), which in yeast is called preautophagosomal structure. 68 The PI3K complex helps to recruit other Atg molecules to IM to elongate the structure. 93
The autophagosome elongation mechanism is built around 2 Ub-like conjugation systems. 74 In one system, the Ub-like protein Atg12 is first activated by Atg7, an E1-like protein, and then transferred by Atg10, an Ub carrier protein–like (E2-like) protein, to Atg5 through a covalent bond. The Atg5–Atg12 complex interacts with Atg16 to form a multimer complex, which is recruited to the IM (the phagophore). In another system, a Ub-like protein, Atg8, or one of its mammalian orthologs, microtubule-associated protein 1 light chain 3 (LC3), is first cleaved by Atg4 to expose the conserved Gly120 at its C-terminus. Atg8/LC3 is then conjugated to phosphatidyl-ethanolamine (PE) via Atg7 and Atg3, another E2-like protein. 38,74 The unconjugated form of Atg8/LC3 (LC3-I) is in the cytosol, whereas the conjugated form (LC3-II) targets the autophagosomal membrane 38 following the Atg5–Atg12–Atg16 complex, which may act like a E3-like enzyme to facilitate the Atg8/LC3–PE conjugation. This association of Atg8/LC3 to the autophagosomes is considered important for the membrane elongation of the phagophore and the eventual enclosure of the membrane to form the autophagosome. The Atg5–Atg12/Atg16 complex may be recycled, whereas Atg8/LC3 could stay on the membrane until it is degraded by the lysosomes.
The intracellular membrane system contributes to autophagosome maturation at multiple levels, probably to guarantee the functionality of the autophagy pathway in various conditions. The autophagosomes fuse with different membranous components as they mature. Evidence from a variety of studies suggests that AV membranes could be contributed by ER, Golgi apparatus, 21 mitochondria 27 and endosomes. The fusion of endosomes and AVs forms vesicles termed amphisomes. 58 Endosomes fuse with AVs at different maturation stages—namely, initial, intermediate, and degradative. 58 However, AVs can fuse with early and late endosomes. 5 Microtubule facilitates fusion of the vesicles by providing the track of movement, 24 and a novel protein, FYCO1, facilitates the trafficking of autophagosomes through microtubule plus end. 78,79 Membrane proteins known to facilitate the fusion process include the Rab-SNARE system, 75 LAMP2, 89 and Beclin 1. 56 In a recent publication, the elongation process of AVs was shown to be independent of Atg5, Atg7, Atg16, and Atg9 by treatment of cytotoxic reagents but dependent on Rab9-positive endosomes and the Golgi network. 71 More remains to be revealed about the mechanisms of AV evolution.
The Proteasome in Health and Disease
UPS plays an important role in maintaining normal cellular functions. Prompt removal of key proteins such as cyclins and certain transcription factors is critical to the precise and timely regulation of intracellular signaling involved in multiple cellular events, including cell growth, transcription, and DNA repair. UPS can regulate cell death through the degradation of some of the key proteins in apoptosis and related pathways, such as p53, I-κB, and inhibitor of apoptosis proteins (IAPs). Hence, inactivation of the proteasome disturbs cellular homeostasis and facilitates the induction of apoptosis. p53 is a tumor-suppressing molecule. Several E3 Ub ligases have been identified to be responsible for p53 degradation. 13 Furthermore, UPS plays an important role in efficient transcription mediated by p53. 107 IAPs, a family of proteins, can also be ubiquitinated and degraded by the proteasome in response to apoptotic stimuli. These proteins can bind to caspases, the key effector proteases of apoptosis, through proteasomes, thus inhibiting apoptosis. 101 Yet, degradation of I-κB by UPS causes the activation of the prosurvival NF-κB pathway.
Impaired proteasome function is found in disease states such as myocardial infarction, 105 neurodegeneration, and alcoholic liver injury. 25 Proteasome inhibitors induce peripheral neuropathy featured by ubiquitinated protein aggregates in vitro and in vivo; this effect is proteasome specific and independent of the chemical scaffold. 14 ER stress exists in diseases caused by proteasome malfunction. It seems that the effect of proteasome inhibitors in causing ER stress and misfolded protein accumulation contributes greatly to cell death. The responsible mechanisms of the proteasome inhibition–induced apoptosis include ER stress, activation of Jun N-terminal kinase (JNK), and inhibition of the NF-κB pathway, leading to the activation of the mitochondrial apoptosis pathways. In our own experiments, we found that proteasome inhibition induces cell apoptosis mainly via ER stress and the mitochondrial apoptosis pathway mediated by Bax and Bak. 17
The Proteasome in Treatment of Cancer
Because of the key regulatory function of the proteasome, proteasome inhibitors have been found to be cytotoxic in vitro for many cancer cells. 1 Thus, inhibition of proteasome has become a novel strategy in cancer therapy. The proteasome inhibitor bortezomib (Velcade) was approved by the Food and Drug Administration in 2003 to treat refractory and relapsed multiple myeloma. Although proteasome inhibitors are effective in the case of myeloma, they have poor efficacy in treating solid tumors in vivo as a single agent.
Recent findings suggest that autophagy suppression in combination with proteasome inhibition can improve the efficacy and selectivity of proteasome inhibitors, serving as a novel strategy of current cancer chemotherapy 18 (see Cooperation of UPS and Autophagy in Cancer). Furthermore, understanding how specific ubiquitination pathways are regulated to identify potential molecular target will help us to define novel molecular targets in cancer therapy. One example is the Ub ligase, which is the type of enzyme that determines substrate selection in the UPS. It is thus crucial to elucidate the substrates and biology regarding these enzymes to find effective drugs against cancer. As a result, some Ub ligases have been identified as potential therapeutic targets. 22 There are several classes of Ub ligases with different catalytic mechanisms, raising both opportunities and challenges in the identification and development of specific small molecule inhibitors.
ER Stress and Cell Death
ER is the site for posttranslational protein modifications, including folding, oligomerization, glycosylation, and disulfide bond formation. ER stress can be caused by the accumulation of misfolded or premature proteins in the ER lumen or the cytosol. 20,28,91 Changes of the environment in the ER lumen, such as the calcium level, the redox status, and the disturbance of ER function (eg, glycosylation and transportation to Golgi complex), could all affect proper protein folding. 12,43 Disturbances in energy metabolism that leads to low ATP production, such as ischemia and hypoxia, also affect protein folding. 52 Misfolding caused by genetic mutations can lead to conformational diseases, such as Huntington disease 51 and Alzheimer disease. 42 Protein misfolding can also be seen in certain infections, as in the transmissible spongiform encephalopathies (the prion disease), 32 or in type II diabetes owing to change of physiochemical parameters in the cellular protein–processing machinery. 30 Many chemical compounds can induce ER stress by disturbing the ER milieu or hampering protein modification and transportation (Table 1 ).
Drugs and Chemicals That Affect Autophagy a

a ER, endoplasmic reticulum
b Approved by the Food and Drug Administration for clinical use.
Prolonged ER stress can lead to decompensation and cell death. 12,84 The major mitigating mechanism for ER stress is the unfolded protein response (UPR), which is mediated by several signaling mechanisms to alleviate ER stress. In mammalian cells, UPR is mediated by PERK, ATF6, and IRE1 pathways 28,91 (Fig. 1 ). PERK phosphorylates eIF2α, which downregulates the synthesis of all proteins except for the transcription factor C/EBP homologous protein (CHOP), also known as growth arrest and DNA damage-inducible gene 153 (GADD153), which is important for ER stress–induced apoptosis. 76 ATF6 upregulates the expression of ER chaperon BiP and the transcription factor XBP-1. As the third arm of UPR, IRE1 activates stress kinases, including JNK and capases, and the processing of the mRNA of Xbp-1, which is important for the production of CHOP/GADD153, BiP, and a variety of ER-associated degradation components to facilitate protein stabilization and degradation. The overall consequence of UPR is the suppression of global protein expression but the upregulation of ER chaperons and proteins involved in degradation pathways. In eukaryotic cells, UPS is the main system to degrade misfolded proteins exporting from the ER. It is the degradation arm in the ER-associated degradation pathway, the mechanism possessed by ER to retrotransport proteins that fail to be modified and/or folded properly and, thus, a major protective mechanism to alleviate ER stress. UPR enhances the proteasomal degradation of unfolded proteins upon ER stress. However, in some cases, ER stress may have inhibitory effects on the UPS. 66
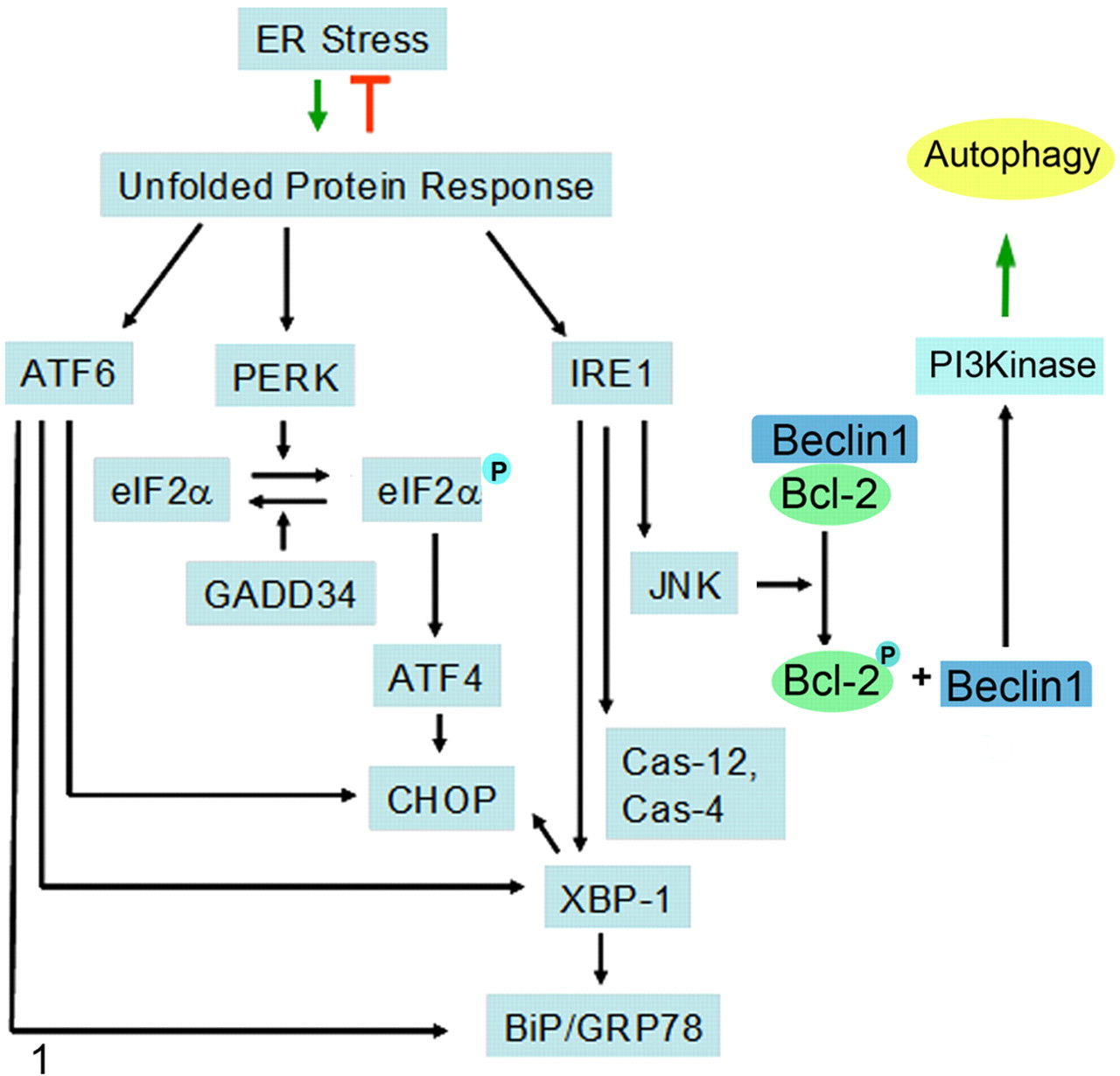
Endoplasmic reticulum (ER) stress and unfolded protein response (UPR). ER stress could be induced by unfolded/misfolded proteins and ER malfunctions. The UPR is activated in 3 separate pathways to promote degradation of the unfolded/misfolded proteins to ease ER stress. PKR-like ER kinase (PERK) is a Ser/Thr kinase that phosphorylates (P) eukaryotic initiation factor 2α (eIF2α), leading to its functional suppression and, therefore, a global shutdown of protein translation, but it allows the translation of a few proteins, including activating transcription factor 4 (ATF4), which upregulates the expression of C/EBP homologous protein (CHOP). ATF6 translocates to Golgi and gets cleaved before it moves to the nucleus. IRE1 can activate transcription factor XBP-1 and stress kinases c-Jun N-terminal protein kinase (JNK). 99 Activated XBP-1, ATF6, and ATF4 collectively lead to the transcriptional activation of a number of genes, including BiP and CHOP/GADD153 (growth arrestand DNA damage-inducible gene 153), to adapt to the stress conditions. JNK can phosphorylate Bcl-2, which would dissociate from the Beclin1/Bcl-2 complex. The increased amounts of freed Beclin 1 can lead to enhanced class III phosphatidylinositol-3-kinase (VPS34/VPS15) activation and, thus, autophagy. As such, JNK potentially links UPR with autophagy activation. 96
As demonstrated in yeast 6,103 and mammalian cells, 53,73 ER stress can activate the other major cellular degradation mechanism, autophagy, which can in turn affect ER stress and, therefore, cell death. The molecular mechanism is known to be in part related to 2 arms of the UPR pathways (Fig. 2 ). Autophagy induced by the expression of the misfolded pathogenic polyglutamine repeats requires the ERK-eIF2α pathway for initiation. 53 The IRE1-JNK pathway is required for the lipid conjugation of LC3 in autophagy induced by ER stress inducers 19,73 and proteasome inhibitors. 19 It seems that ATF6 is not required for ER stress–induced autophagy. 73 Autophagy in response to ER stress can remove polyubiquitinated proteins, rendering cells more resistant to ER stress. 19,73
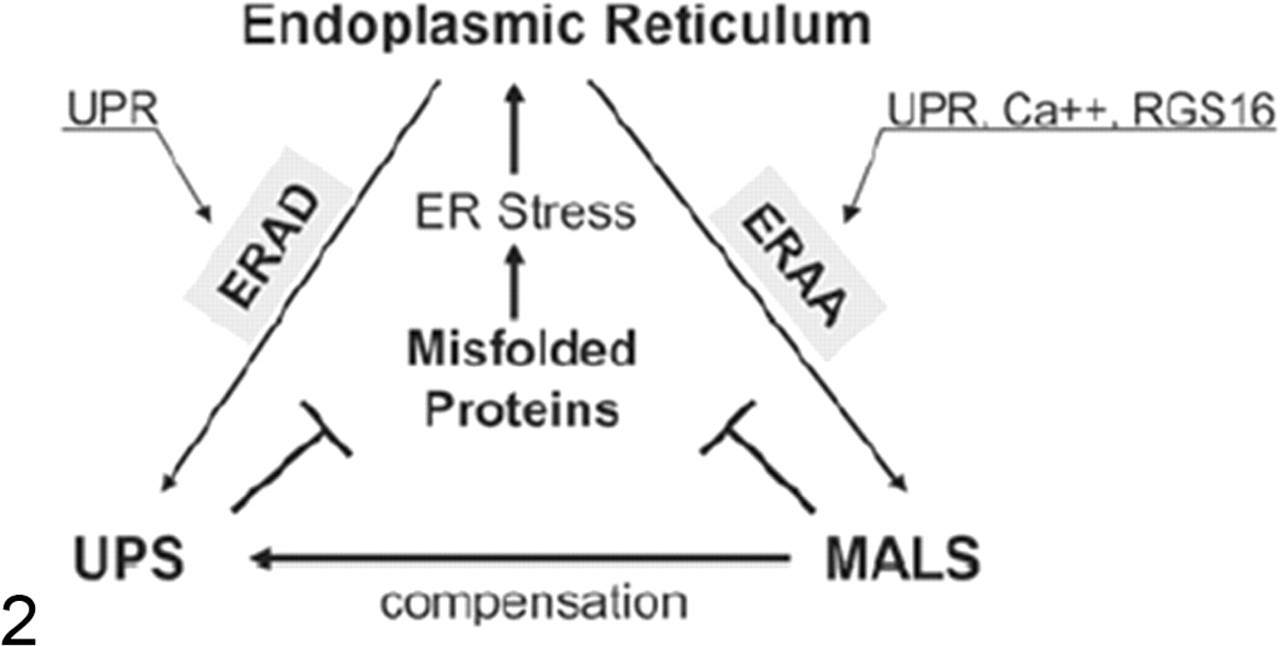
The endoplasmic reticulum (ER) commands the degradation of misfolded proteins by the proteasome and autophagy. Misfolded proteins are degraded by the ubiquitin–proteasome system (UPS) and the macroautophagy-lysosome system (MALS). ER stress activates proteasome degradation by the ER-associated degradation (ERAD) pathway, which is mediated by the unfolded protein response (UPR) involving all 3 branches of ATF6, PERK, and IRE1 (see Figure 1). Yet, ER stress activates MALS by the ER-activated autophagy (ERAA) pathway, which is mediated by a limited UPR involving PERK and/or IRE1 and UPR-independent mechanisms. ER calcium leakage is a major UPR-independent mechanism. In addition, molecules regulating G protein signaling, such as RGS16, have been implicated in the activation of ERAA.
Another mechanism by which ER stress reduces autophagy is calcium mediated (Fig. 2). 33 As the major cellular calcium reservoir, ER can release increased amount of calcium to the cytosol. Calcium signaling is independent of the UPR mediated by IRE1, ATF6, and PERK 7 and may lead to the suppression of the mTOR via CaMMKβ, which activates AMPK. In addition, the initiation of autophagy requires class III PI3K, and calcium plays an important role in class II PI3K signaling. 59,97 Whether calcium is also important for class III PI3K activation remains to be determined.
Interestingly, ER has been proposed to be an initiation site of autophagy. 37,87 Some ER components can be found on autophagosomes. 34 One recent study showed that DFCP1, an ER residential PI3P-binding protein, colocalizes with autophagosome markers. 3 Electron microscopic tomograph indicated that AV membranes could be linked to ER. 29,102 Other candidates of the membrane source of nascent AVs include the Golgi complex, 47,71 mitochondria, 27,61 and plasma membrane. 86 A recent publication showed that mitochondria outer membrane is associated with autophagosomal marker proteins Atg5 and LC3 under starvation and that the formation of the autophagosome depends on ER–mitochondrion lipid transfer. 27 More conclusive evidence to substantiate these findings remains to be unveiled.
Cooperation of UPS and Autophagy in Cancer
At its basal level, autophagy contributes to the cellular homeostasis and plays an important role in embryonic development. 67 Its activity can be elevated in response to a variety of physiologic or pathologic stimuli to sustain normal cellular function—for example, to provide cells with sufficient nutrients during starvation. Yet, successful removal of misfolded proteins and malfunctional organelles helps cells resist death. Autophagy can mitigate oxidative stress via removal of damaged mitochondria, a key source of reactive oxygen species (ROS); 45 it is also important for the innate immunity by participating in the clearance of certain pathogens. 54 Thus, autophagy is known to be mainly a prosurvival mechanism. 40,65
Nevertheless, autophagy can be associated with cell death. 4,15 For example, in Drosophila salivary gland, induction of autophagy contributes to premature cell death in a caspase-dependent manner. 8 There are several theories about how autophagy may induce cell death: overdigestion of cytoplasmic proteins, activation of apoptotic pathway, and selective digestion of cytoprotective factors. 104 The contradictory role of autophagy in cell survival and death can depend on factors such as the level of autophagy and the status of the cell. We have found that cells at different transformation states respond differently to ER stress-induced autophagy, which is more protective to transformed cancer cells than to normal or nontransformed cells. 18 Such differential response has clinical implications in developing novel anticancer treatment strategies. The challenge is to understand the underlying mechanism of how autophagy regulates cell death in different conditions for maximal benefits.
Cancer cells apply UPS and autophagy pathways to adapt to the fast proliferation status and to escape apoptotic and necrotic cell death. Hence, UPS and autophagy are mostly regarded as prosurvival mechanisms in developed cancer cells. Oncogenically transformed cells may have a stronger ability to initiate autophagy and become more dependent on this mechanism to survive. 17,63 It is possible that tumor cells have a higher metabolic rate and require a higher level of energy supply such that autophagy serves as an efficient way to provide the additional source of energy and compositional building blocks. Another possibility is that autophagy promotes the removal of misfolded proteins, which may more easily accumulate in cancer cells. 19
The mammalian homolog of Atg6, Beclin1, is important in autophagic control of tumorigenesis. Beclin1 forms a complex with VPS34, VPS15, and Atg14 to function as a key initiation mechanism. VPS34 is a type III PI3K required for autophagy. Although the mechanism of this complex is not known yet, deletion of Atg6/Beclin1 in yeast or mammalian cells leads to the blockage of autophagy, likely at an upstream point. 70,83 Bcl-2 and Bcl-xL are antideath Bcl-2 family proteins, but they can also bind to Beclin1 and suppress its function. 55,80 Conversely, Beclin1 may inhibit their functions because it binds to them like a BH3-only prodeath molecule. 11,23,62,72,80 Although Beclin1-null mice are embryonic lethal, Beclin1 heterozygous mice are normal at the beginning but develop multiple tumors later on, suggesting that Beclin1 is a haploinsufficiency tumor suppressor. 83 Loss of the heterozygosity of Beclin1 is frequently seen in breast and ovarian cancers. 57 Defects in autophagy are thought to promote oncogenesis in normal cells owing to failure to degrade malfunctional proteins and increase of genomic instability caused by ROS damage. 63 –65 Mitochondria are the main source of ROS generation. Autophagy can maintain cellular homeostasis by removing damaged organelles, including mitochondria, a process named mitophagy, which has been well characterized and recently reviewed. 39 As the consequence, autophagy may suppress tumorigenesis by mitigating ROS and genome damage. 40,65 Alternatively, there is evidence that p62, which binds to Ub and LC3, serves as a signal for selective degradation by autophagy. 9,49,77 Suppression of autophagy causes accumulation of p62, which is tumorigenic through the activation of NF-κB. 64
The role of autophagy in protecting existing cancer cells during stress has also been well documented. When cancer cells are exposed to radiation or chemotherapeutic agents, autophagy is activated. 26,36,50,60 By activating autophagy, tumor cells could better cope with the stress for survival. 16,18,36 Consequently, suppression of autophagy leads to higher levels of cell death. 2,16,18,26 As a developing strategy in cancer treatment, combined use of chloroquine (which suppresses autophagy by impairing lysosomal pH) and cytotoxic reagents can result in better therapeutic effects in animal tumor models as well as tumor cell lines. 2,16,18,33,36,38,61 Further understanding of the initiation and regulation mechanism of autophagy will help to identify new drugable targets.
It is now a well-established notion that autophagy and UPS are functionally connected, a connection that especially pronounced under ER stress (Fig. 2). Mouse strain with conditional knockout of Atg7 or Atg5 shows neurodegeneration with Ub-positive pathology. 48,69 Inhibition of proteasomes increases cell death by causing ER stress and subsequently turns on autophagy as a mitigating mechanism. 18,19 Both UPS and autophagy can be activated in response to ER stress caused by misfolded proteins. Compensatory activation of autophagy during UPS inhibition is also mediated by the misfolded protein-induced ER stress (Fig. 2). The compensatory effect of the proteasome during autophagy inhibition has not been reported, suggesting that proteasome degradation is a more limited mechanism for clearing misfolded proteins. The underlying molecular mechanism of autophagy initiation following proteasome inhibition is discussed above (see ER Stress and Cell Death). Besides the p62-mediated selection mechanism, autophagy is dependent on histone deacetylase 6 (HDAC6). 77 HDAC6 can bind to polyubiquitinated misfolded proteins, 44 and it is required for autophagic degradation of aggregated mutant huntingtin in cultured cells 35 and the mutant androgen receptor in a model of Kennedy disease. 77 The Ub recognition ability of p62 and HDAC6 renders them the ability to degrade the aggregated proteins.
Promotion of apoptosis by proteasome inhibition, as discussed earlier, is being practiced in cancer treatment. 22 However, proteasome inhibitors have not been found to be effective in treating solid tumors in clinic. Although there are many possible reasons, activation of the protective autophagy could be a critical one. 19,106 For example, autophagy induced by proteasome inhibitors could lessen the degree of ER stress, which could be responsible for its protective effects against cell death. 19 These effects are consistent with the evidence of autophagy serving as a prosurvival mechanism in cancer cells. 16,36,60,63 Suppressing autophagy could thus be an effective strategy to enhance the killing of cancer cells of different origins other than the multiple myeloma by proteasome inhibitors. Simultaneous inhibition of autophagy and proteasome function can be more effective and advantageous, also because of the low toxicity for normal cells. 18
Summary
The 2 synergistic degradation systems in eukaryotic cells, UPS and autophagy–lysosome system, are cooperative and complementary to maintain cellular homeostasis. They can be coactivated to degrade misfolded proteins, as in the case of degradation of the Z variant of human α-1 antitrypsin. 94 There are also compensatory effects when one is dysfunctional (Fig. 2). They both can play vital roles in alleviation of ER stress. As a versatile degradation system—in terms of the type of targets and the substrate delivery mechanism—autophagy can help the cell to survive through a variety of stress conditions.
Recent findings about the increased autophagic activity upon proteasome inhibition during ER stress have shed light in development of novel cancer therapy strategies. Autophagy seems to be more beneficial for cancer cells to survive through stressful conditions, including ischemia, hypoxia, chemotherapy, and radiation therapy. 50 Further understanding of the molecular mechanism—that is, the communication between autophagy and the proteasome in response to different stimuli—will guide pharmaceutic development of novel treatment methods. Combinational use of agents acting through different targets can be more effective for cancer therapy. Autophagy-inducing and autophagy-suppressing drugs can be used with traditional cytotoxic agents, depending on the scenario. The coordination of autophagy and UPS during ER stress will remain as challenging yet inspiring topics not just in cancer but in many other diseases.
Footnotes
The authors declared no potential conflicts of interests with respect to the authorship and/or publication of this article.
The authors disclosed receipt of the following financial support for the research and/or authorship of this article: Dr X.-M. Yin is in part supported by National Institutes of Health grants (CA83817, CA111456).