
Abstract
The pathogenesis of lower respiratory tract disease from the pandemic 2009 H1N1 (H1N1v) influenza A virus is poorly understood. Therefore, either H1N1v virus or a seasonal human H1N1 influenza A virus was inoculated into cynomolgus macaques as a nonhuman primate model of influenza pneumonia, and virological, pathological, and microarray analyses were performed. Macaques in the H1N1v group had virus-associated diffuse alveolar damage involving both type I and type II alveolar epithelial cells and affecting an average of 16% of the lung area. In comparison, macaques in the seasonal H1N1 group had milder pulmonary lesions. H1N1v virus tended to be reisolated from more locations in the respiratory tract and at higher titers than seasonal H1N1 virus. In contrast, differential expression of messenger RNA transcripts between H1N1v and seasonal H1N1 groups did not show significant differences. The most upregulated genes in H1N1v lung samples with lesions belonged to the innate immune response and proinflammatory pathways and correlated with histopathological results. Our results demonstrate that the H1N1v virus infects alveolar epithelial cells and causes diffuse alveolar damage in a nonhuman primate model. Its higher pathogenicity compared with a seasonal H1N1 virus may be explained in part by higher replication in the lower respiratory tract.
Keywords
The newly emerged influenza A virus of subtype H1N1 was declared pandemic as of June 11, 2009, and as of November 22 more than 622,482 confirmed cases had been reported worldwide (http://www.who.int/csr/don/2009_11_27a/en/index.html). The new variant of the H1N1 subtype (H1N1v virus), probably originating from a swine reservoir 6 , has caused at least 7,826 confirmed deaths, mostly with a clinical diagnosis of atypical pneumonia or acute respiratory distress syndrome. 15 Influenza viruses may vary substantially in the human disease they cause. 22,24 The pathogenesis of H1N1v virus infection in the human respiratory tract is largely unknown, as is the resultant damage compared with that caused by seasonal influenza viruses, which are well-adapted to their human host.
To establish a nonhuman primate model of influenza pneumonia, we inoculated either the H1N1v virus or a seasonal human H1N1 influenza A virus (seasonal H1N1 virus) into cynomolgus macaques and performed virological, pathological, and microarray analyses. The cynomolgus macaque is a suitable species to model human pneumonia from infection by respiratory viruses, including HMPV, 14 SARS virus, 7 and influenza viruses. 1,12,13,20
Methods
Virus Preparation
Influenza virus A/Netherlands/602/09 (H1N1v virus) was isolated from a patient who had traveled from Mexico to the Netherlands and was the first laboratory-confirmed case of H1N1v virus infection in the Netherlands. This virus was cultured from a nasopharyngeal swab in 11-day-old embryonated chicken eggs and subsequently passaged once in Madin-Darby canine kidney (MDCK) cells. The sequence of the virus that was used for inoculation of macaques was identical to that of the clinical isolate. This virus differs in 8 amino acid positions from influenza virus A/California/4/2009 (P100S, T214A and I338V in HA, I108V and V407I in NA, T373I in NP, P224S and M581L in PA; GISAID accession numbers EPI178246-250, EPI178467, EPI178290, and EPI178291). None of the differences map to known pathogenicity markers of influenza A virus. In fact, A/Netherlands/602/2009 is more representative (ie, more consensus-like) than A/California/4/09; most amino acid changes in A/Netherlands/602/2009 compared with A/California/4/2009 were also found in the majority of sequenced strains in the database. The only unique sequence changes in A/Netherlands/602/2009 are I108V and V407I in neuraminidase, which can be considered conservative changes, not previously associated with virulence. Until now, the H1N1v viruses have shown low genetic diversity and we consider the isolate used to be representative of the currently circulating H1N1v virus. The seasonal H1N1 influenza virus A/Netherlands/26/07 (seasonal H1N1 virus) was isolated from a patient during the 2006–2007 influenza season and was subsequently passaged 3 times in MDCK cells.
Experimental Protocol
Experiments were performed under biosafety level 3 conditions under an animal study protocol approved by the Institutional Animal Welfare Committee. Two weeks prior to infection, temperature loggers (DST micro-T ultra small temperature logger; Star-Oddi) that recorded the body temperature every 15 minutes were placed in the peritoneal cavity of cynomolgus macaques under ketamine and domitor (medetomidine hydrochloride) anesthesia. Two groups of 4 macaques were inoculated with 1 × 107 median tissue culture infectious dose (TCID50) of seasonal H1N1 virus or H1N1v virus in 5 ml of phosphate-buffered saline (PBS) (4 ml intratracheally and 1 ml intranasally) under ketamine and domitor anesthesia. Swabs were collected daily from macaques under ketamine anesthesia from nose and throat and placed in 1 ml of transport medium. 5 All macaques were euthanatized 4 days after inoculation by exsanguination under ketamine anesthesia and were necropsied according to a standard protocol. Randomly collected lung tissue sections from 4 PBS (sham) inoculated age-matched cynomolgus macaques were used as control samples for below analyses.
Virus Titration
Virus titers were determined in nose, throat, and rectum swabs and in the tissues collected during necropsy: nasal turbinate, tonsil, tracheobronchial lymph node, trachea, bronchus, lung, spleen, liver, kidney, jejunum, colon, duodenum, heart, eyelid, pancreas, and brain. Tissue samples were weighed and subsequently homogenized with a FastPrep homogenizer (MP Biomedicals Europe, Illkirch, France) in 3 ml of transport medium. For lung, 4 samples of lung tissue collected from standard locations (cranioventral, craniodorsal, caudoventral, and caudodorsal) in the right lung were pooled for each animal. Virus titrations were performed by end point titration in MDCK cells. MDCK cells were inoculated with 10-fold serial dilutions of homogenized tissues, nose, throat, or rectum swabs. One hour after inoculation, cells were washed once with PBS and grown in 200 μl of infection media, consisting of EMEM (Lonza, Breda, The Netherlands) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM glutamine, 1.5 mg/ml sodium bicarbonate (Cambrex), 10 mM HEPES (Lonza), nonessential amino acids (MP Biomedicals Europe), and 20 μg/ml trypsin (Cambrex). Three days after inoculation, the supernatants of infected cell cultures were tested for agglutinating activity using turkey erythrocytes as an indicator of infection of the cells. Infectious titers were calculated from 5 (homogenized tissues) or 4 (swabs) replicates by the method of Spearman-Karber. 10
Histopathological Examination and Immunohistochemistry
Samples for histological examination were stored in 10% neutral-buffered formalin (lungs after inflation with formalin), embedded in paraffin, sectioned at 4 μm, and stained with hematoxylin and eosin (HE) for examination by light microscopy. Selected lung sections also were stained with periodic acid–Schiff (PAS) for detection of mucoid substances. The following tissues were examined by light microscopy: left lung (cranial, medial, and caudal lobe), nose, nasal turbinates, nasal septum, larynx, trachea, bronchi, tracheobronchial lymph node, eyelid, tonsil, heart, liver, spleen, kidney, pancreas, duodenum, jejunum, colon, adrenal gland, and brain. Semiquantitative assessment of influenza A virus–associated inflammation in the lung was performed as reported earlier. 7 Briefly, each slide was examined for inflammatory foci at 10× objective, and each focus was scored for size and severity of inflammation in different areas of the lung. For the size of the inflammatory focus in the alveoli, we used 1, smaller or equal than area of 10× objective; 2, larger than area of 10× objective and smaller than area of 2× objective; and 3, larger than area of 2× objective. For the severity of inflammation, we scored 1, mild (few inflammatory cells); 2, moderate (moderate numbers of inflammatory cells and/or mild necrosis and edema with erythrocytes); and 3, marked (many inflammatory cells and/or necrosis and edema with inflammatory cells). Slides were examined without knowledge of the identity of the animals. The cumulative scores for the inflammatory foci provided the total score per animal.
Sequential slides were used for detection of influenza A virus nucleoprotein in tissues as described before. 20 Endogenous peroxidase was blocked with 3% hydrogen peroxide. Lung sections from a domestic cat with experimental avian H5N1 influenza virus infection were used as positive controls. Semiquantitative assessment of influenza virus antigen expression in the lungs was performed as follows: for assessment of alveoli, 100 arbitrarily chosen 20× objective fields of lung parenchyma in all lung sections were examined by light microscopy for the presence of influenza virus antigen expression without the knowledge of the identity of the animals. The scores for each animal were presented as number of positive fields per 100 fields (%).
For detection of pankeratin in lung tissues of an H1N1v infected macaque, 3-μm formalin-fixed, paraffin-embedded sections were deparaffinized and rehydrated to distilled water. Antigen retrieval was performed at 100°C for 15 minutes with citric acid buffer pH 6.0. Slides were cooled for 20 minutes on ice in the same solution. After washing with PBS, endogenous peroxidase was blocked with 3% hydrogen peroxidase. The slides were briefly washed with 0.05% PBS Tween 20 and incubated with a mouse IgG1 antibody against human pankeratin AE1/AE3 (Neomarkers, Fremont, CA) (2 μg/ml) or mouse IgG1 isotypecontrol (R&D Systems Europe, Abingdon, UK) (2 μg/ml) in PBS/0.1% BSA for 1 hours at room temperature. After washing, sections were incubated with horseradish peroxidase-labeled goat-anti-mouse IgG1 (Southern Biotech) (5 µg/ml) in PBS/0.1% BSA for 1 hour at room temperature. Horseradish peroxidase activity was revealed by incubating slides in 3-amino-9-ethylcarbazole (AEC) (Sigma) for 10 minutes, resulting in bright red precipitate, followed by counterstaining with hematoxylin.
Microarrays
One sample of lung tissue without macroscopically visible pathologic changes (nonlesional) and—if present—1 sample of lung tissue with macroscopically visible pathologic changes (lesional) were randomly selected from the right lung of each animal. Lesional lung samples for microarray were only available from 1 of 4 macaques infected with seasonal H1N1 virus because of the limited area of lung affected. Also, 1 of 4 nonlesional lung samples from macaques infected with H1N1v virus was not retained for analysis. Lung tissue samples were collected in RNAlater (Ambion), and total RNA was isolated and purified using Trizol Reagent (Invitrogen) and the RNEasy mini kit (Qiagen), respectively. RNA was subsequently labeled using the One-Cycle Target Labeling kit (Affymetrix) and hybridized onto Affymetrix GeneChip Rhesus Macaque Genome Arrays (Affymetrix), according to the manufacturer’s recommendations. Image analysis was performed using Gene Chip Operating Software (Affymetrix). Data was preprocessed for background correction and normalized using variance stabilization. 8 Transformed probe values were summarized into 1 value per probe set by the median polish method (part of the Robust Multiarray Averaging [RMA] method). Probe set-wise comparisons between the experimental conditions were performed by limma, 21 which is similar to analysis of variance. Correction for multiple testing was achieved by requiring a false discovery rate (FDR) of 0.05, calculated with the Benjamini-Hochberg procedure. Further downstream processing and interpretation of data including cluster analysis, visualization, and pathway analysis were performed using Spotfire DecisionSite 9.1 for Functional Genomics (Spotfire Inc) and Ingenuity Pathways Knowledge Base (Ingenuity Inc). Hierarchical cluster analysis (single linkage) was performed with a set of transcripts identified as being differentially regulated in at least 1 of the 6 direct comparisons with a FDR ≤ 0.05 and 2log-fold change ≥ 1 threshold.
Statistical Analysis
Data were compared using the Mann-Whitney test (affected lung area, histopathology score, percentage virus-infected cells, virus titers in different postmortem tissues). Differences were considered significant at P < .05.
Results
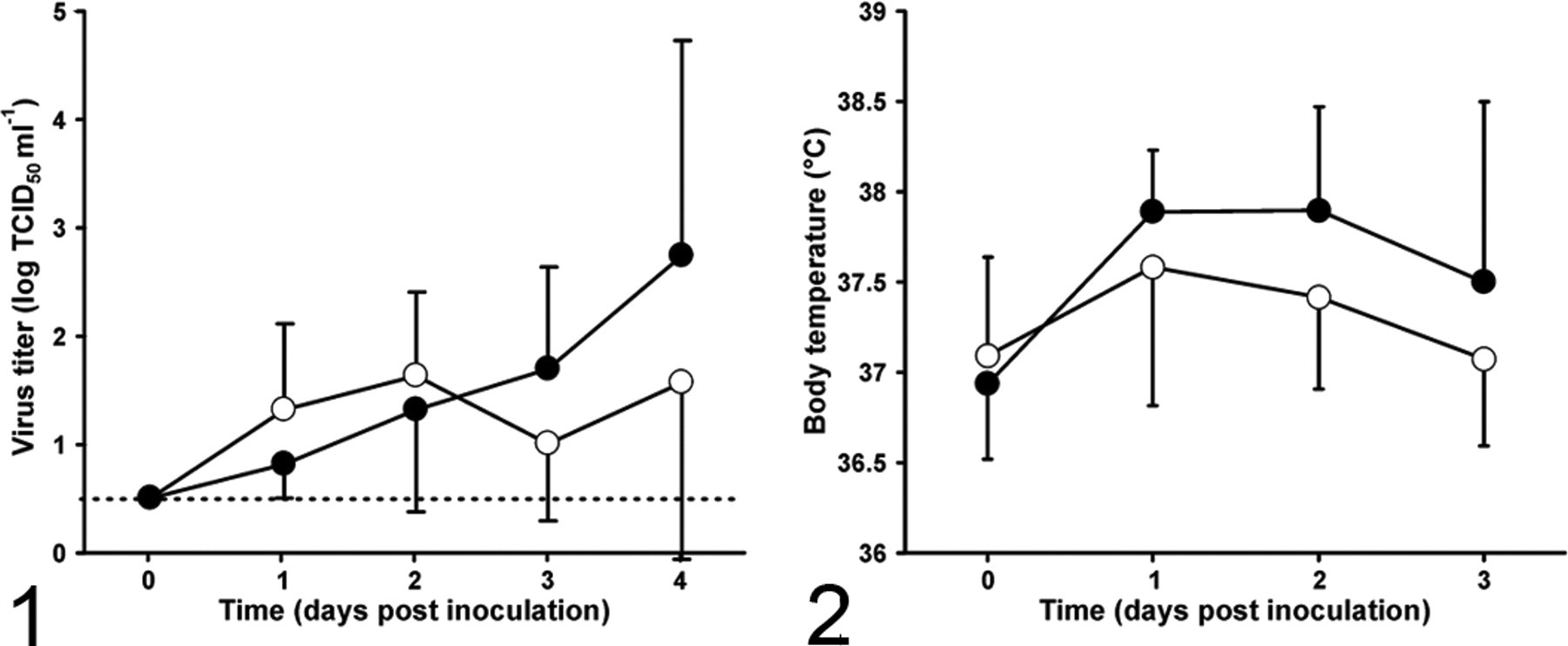
Upon experimental inoculation with the H1N1v virus, viral loads in throat swabs increased progressively in the H1N1v group between 1 and 4 days post inoculation (dpi), when the mean titer was 2.8 log TCID50 per milliliter (Fig. 1). In contrast, viral loads peaked on 2 dpi at a lower level of 1.6 log TCID50 per milliliter in the seasonal H1N1 group. Virus was detected sporadically in nasal swabs of animals from both groups and could only be demonstrated in rectal swabs of 1 animal in each group at 4 dpi. The body temperature in the H1N1v group peaked at 1 dpi and decreased in the following 2 days (Fig. 2). The body temperature in the seasonal H1N1 group followed a similar trend but peaked at about 0.3°C lower. One of the macaques from the H1N1v group was lethargic at 4 dpi.
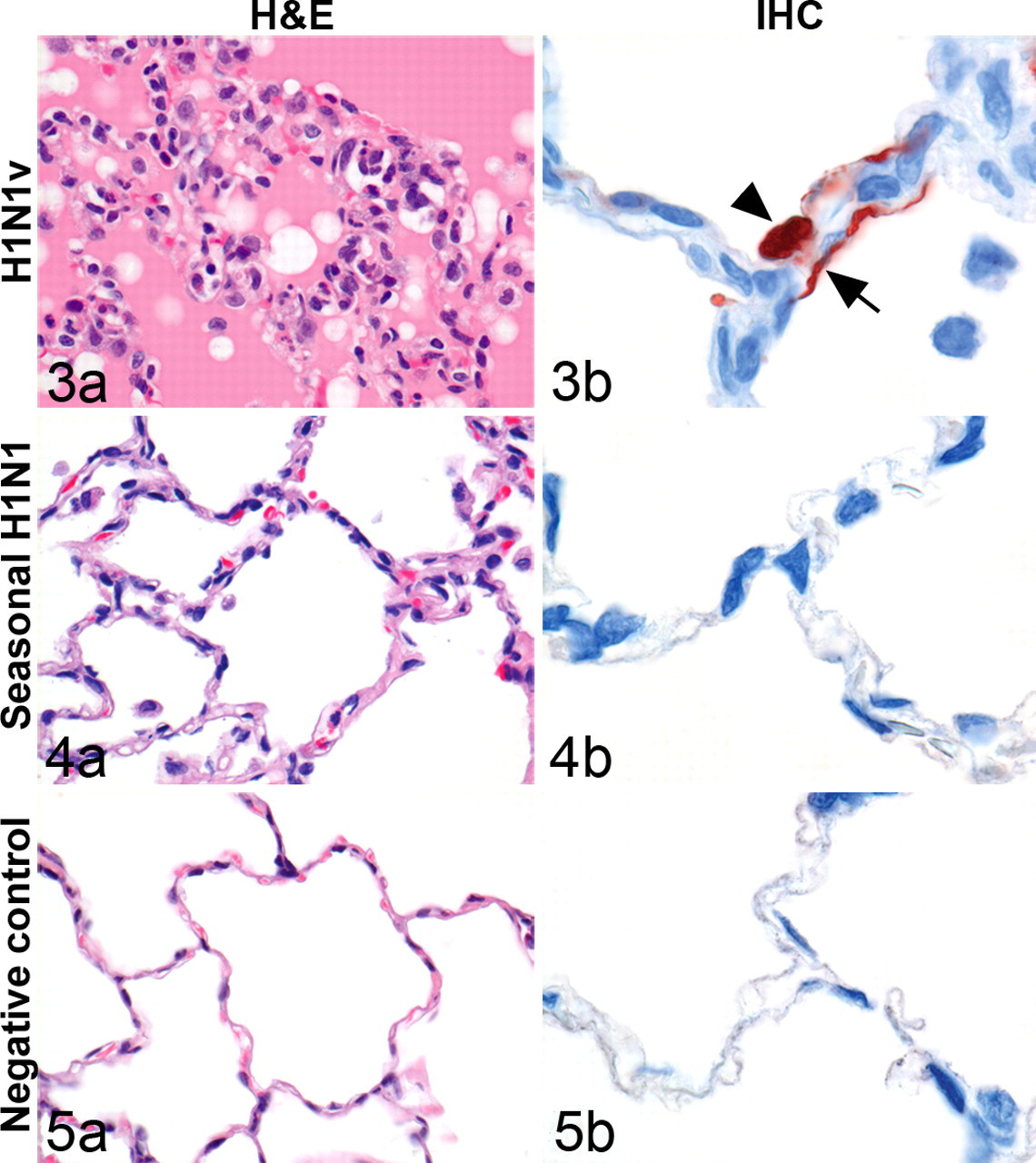
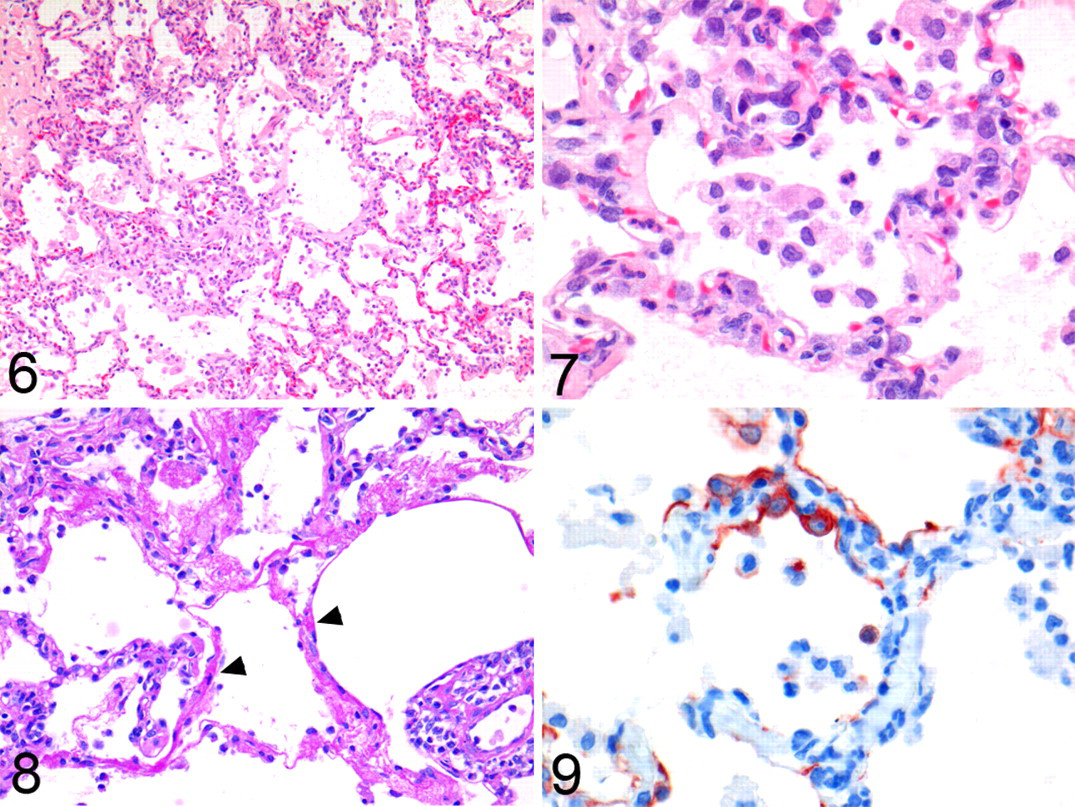
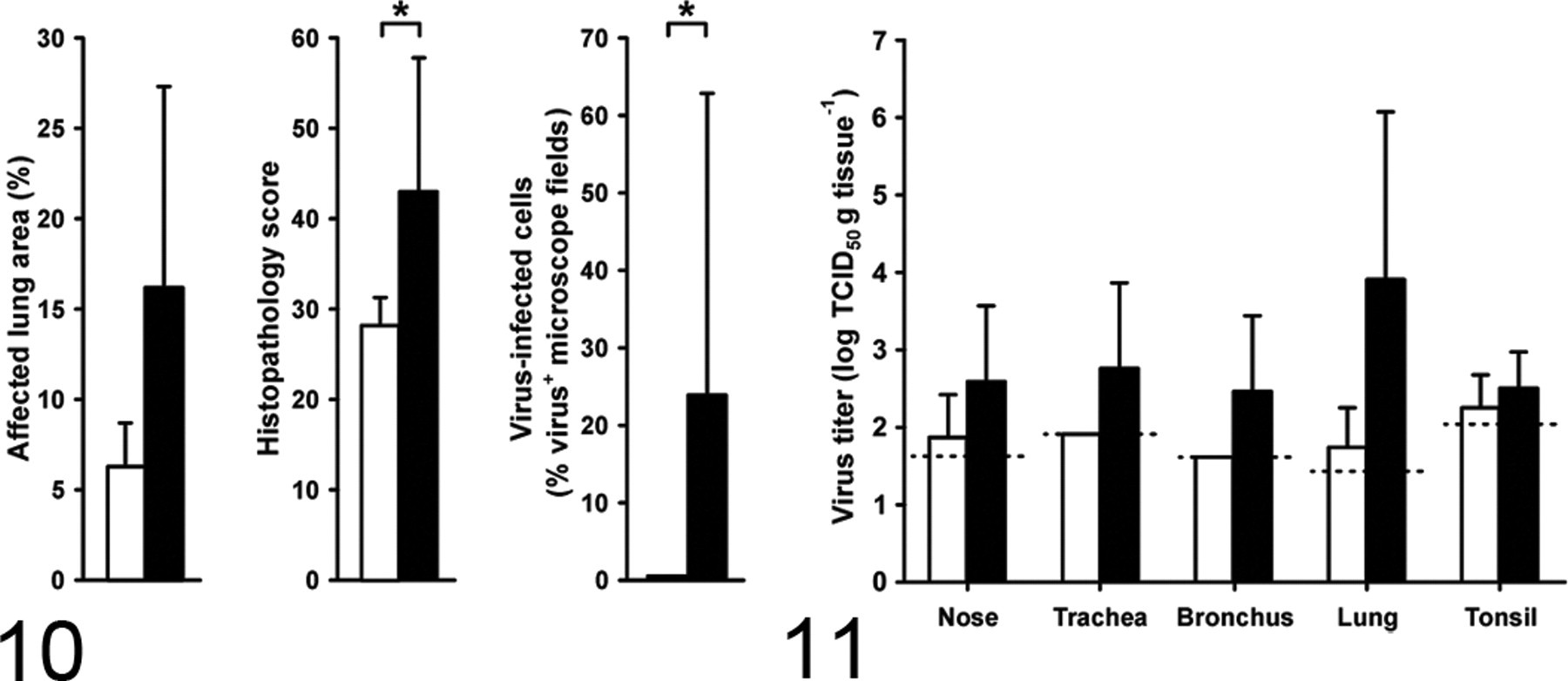
On necropsy at 4 dpi, the macaques in the H1N1v group had focal or multifocal pulmonary consolidation and enlarged tracheobronchial lymph nodes. No lesions were seen in the extrarespiratory tissues. Histologically, pulmonary consolidation corresponded with diffuse alveolar damage (DAD) (Figs. 3-5). This DAD was characterized by flooding of highly proteinaceous fluid into alveolar lumina (Figs. 3a, 6 and 7) with rare PAS-positive hyaline membranes (Fig. 8), necrosis of alveolar epithelial cells, and accumulation of many neutrophils and alveolar macrophages (Figs. 3a, 6, 7). Necrosis of alveolar epithelial cells was demonstrated by a partial loss of pankeratin-expressing pneumocytes (Fig. 9). Other lesions were moderate bronchiolitis, characterized by epithelial necrosis and peribronchiolar infiltration with moderate numbers of macrophages and lymphocytes and few plasma cells, neutrophils, and eosinophils. There were mild bronchitis and tracheitis, with epithelial necrosis and moderate numbers of neutrophils, macrophages, and lymphocytes and a few plasma cells and eosinophils in the lamina propria and submucosa. In addition, there was a mild rhinitis, characterized by mild epithelial necrosis and a few neutrophils, macrophages, and lymphocytes, and there were rare plasma cells and eosinophils in the lamina propria. The macaques in the seasonal H1N1 group had significantly fewer extensive pulmonary lesions than in the H1N1v group (Fig. 10). The alveoli had no flooding with edema fluid, only mild necrosis and fewer inflammatory cells. These inflammatory cells were a few neutrophils, macrophages, lymphocytes, and plasma cells that were located predominantly in the alveolar septa. Neither group had histological lesions in any of the extrarespiratory tissues.
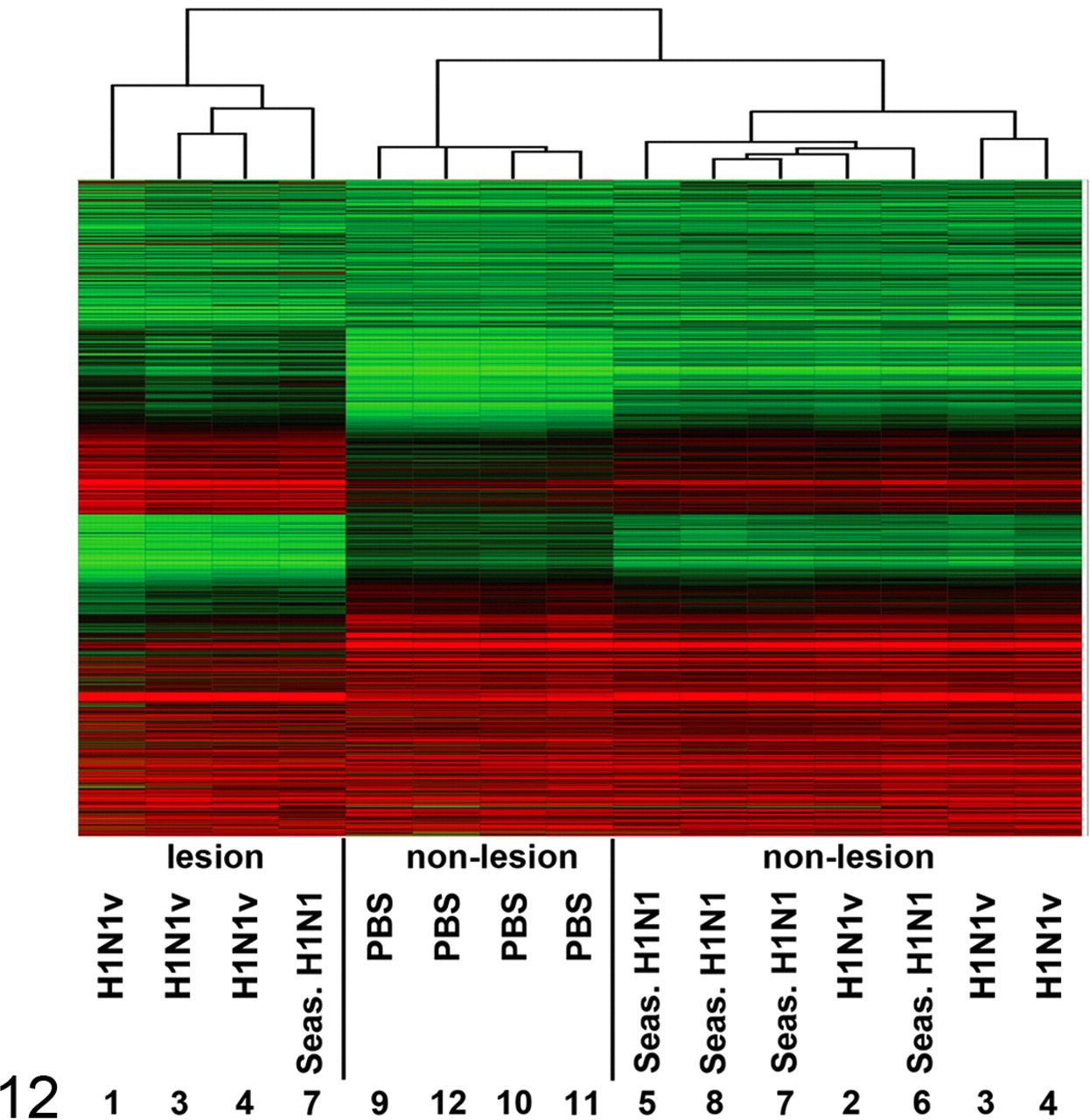
Cluster analysis of gene expression profiles from lesional and nonlesional lung samples from macaques infected with H1N1v virus or seasonal H1N1 virus. Hierarchical clustering of 15 gene expression profiles reveals 3 clusters. Normalized absolute hybridization signals ranged from 24 (green) to intermediate (28, black) and peaked at 213 (red).
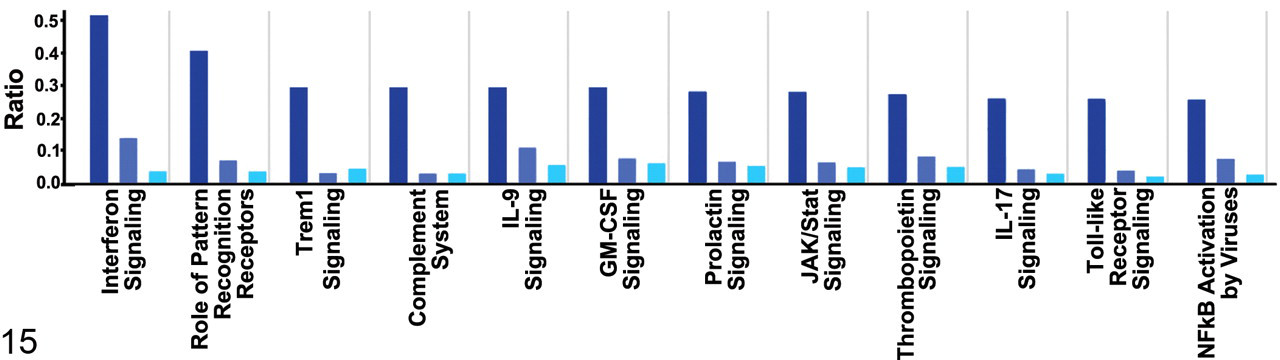
Top 12 most significant (largest P value < 10-6) and relevant differentially expressed molecular pathways for H1N1v lesional samples (Ingenuity Pathways Knowledge Base). The y-axis depicts the fraction (ratio) of the genes assigned to a particular pathway that were differentially regulated (limma analysis, FDR ≤ 0.05, 2log-fold change ≥1). The dark, intermediate, and light blue bars refer to the comparative pathway analysis of H1N1v lesional samples, H1N1v nonlesional samples, and seasonal H1N1 nonlesional samples with control samples.
By immunohistochemistry, all 4 macaques in the H1N1v group expressed influenza virus antigen in alveolar epithelial cells, both type I pneumocytes (77 of 100 positive cells counted) and type II pneumocytes (23 of 100 positive cells counted), in or at the edges of the alveolar lesions (Fig. 3b). Except for a few epithelial cells in larynx and nasal septum of 1 macaque, influenza virus antigen expression was absent in other tissues in the H1N1v group. There was no expression of influenza virus antigen in any tissues of the seasonal H1N1 group (Figs. 4a). Neither lesions nor expression of influenza virus antigen were observed in sham-inoculated macaque lungs (Figs. 5a and 5b). The macaques in the seasonal H1N1 group had significantly fewer severe pulmonary lesions (Fig. 6), lacking edema fluid, with less necrosis and fewer inflammatory cells.
Virus was isolated from the respiratory tract and tonsils, whereas other tissues tested negative. In the H1N1v group, virus was isolated from all 4 macaques, and positive tissues were nose, trachea, bronchus, lung, and tonsil. In the seasonal H1N1 group, virus was isolated from 2 of 4 macaques, and positive tissues were nose, lung, and tonsil. Viral titers, particularly in the lungs, tended to be higher in the H1N1v group than in the seasonal H1N1 group (Fig. 11).
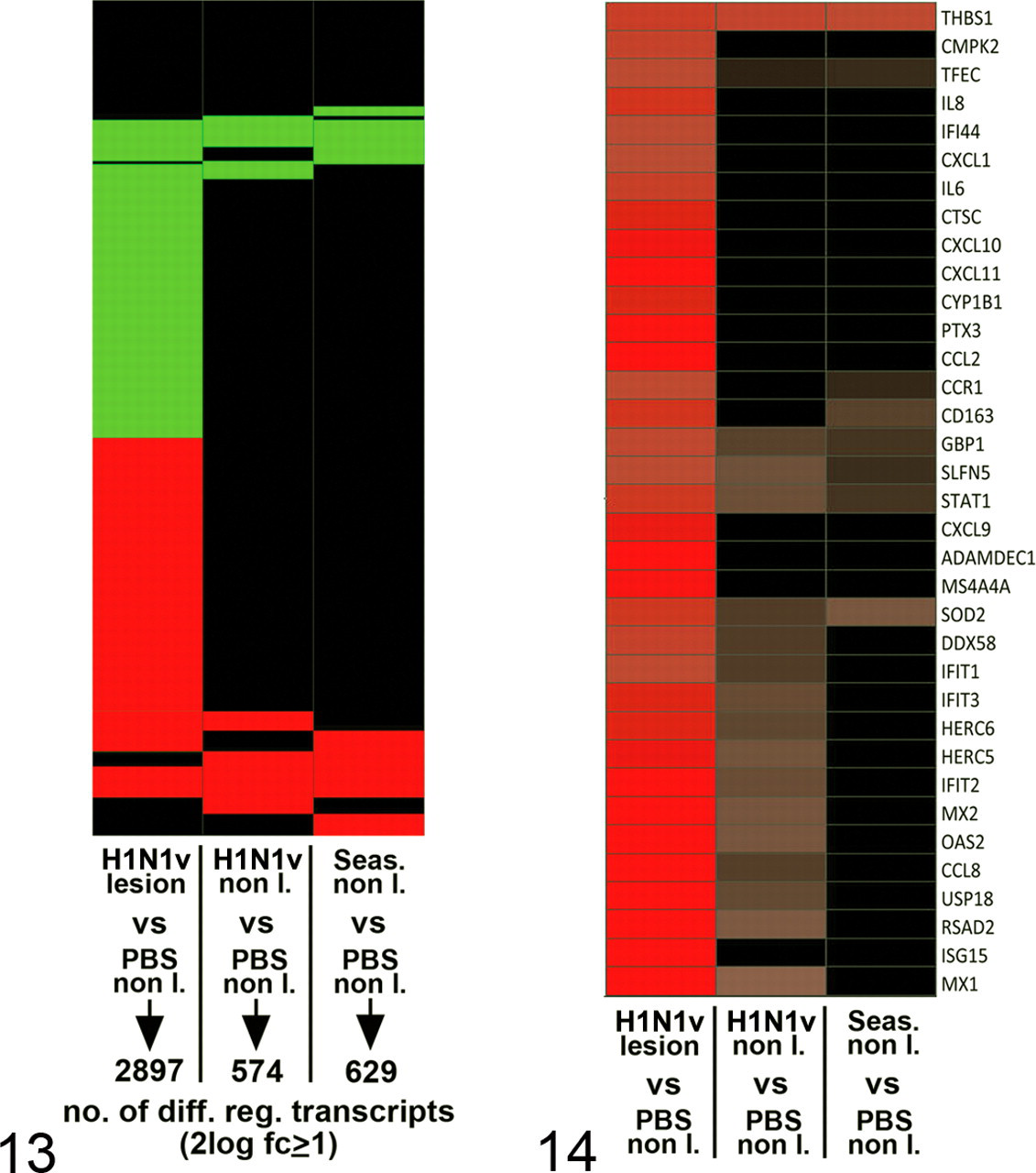
Microarray analysis followed by hierarchical clustering of messenger RNA (mRNA) expression profiles revealed 3 clusters: samples of sham-infected lungs (controls), samples of lungs without lesions (nonlesional), and samples of lungs with lesions (lesional) (Fig. 12). Cluster analysis did not separate samples of the H1N1v and the seasonal H1N1 groups. Limma analysis 21 identified almost 3,000 mRNA transcripts differentially expressed between H1N1v lesional and control samples (FDR ≤ 0.05 and a 2log-fold change ≥ 1) and about 600—which largely overlapped—between nonlesional (both H1N1v and seasonal H1N1) and control samples (Fig. 13). About 1,500 transcripts between lesional and nonlesional (both H1N1v and seasonal H1N1) samples were differentially expressed but only 3 between nonlesional samples of the H1N1v and seasonal H1N1 groups (data not shown). Collectively, cluster and limma analyses showed that lung samples with the same gross pathology exhibited virtually identical mRNA expression profiles, lesional samples differing significantly from nonlesional ones.
The top 35 upregulated genes in H1N1v lesional samples mostly belonged to innate immune response and proinflammatory pathways (Fig. 14) and correlated with histopathology. For example, the high mRNA expression for CCL2, CCL3, CCL8, CXC10, IL8, and CXCL1—known chemoattractants for neutrophils and monocytes—corresponded with the abundance of these cell types in lesional samples (Fig. 3a). Pathway analysis (Ingenuity) of the differentially expressed genes showed profiles (Fig. 15) similar to those observed previously in macaque influenza virus infections. 4
Discussion
We here show that the H1N1v virus is found throughout the respiratory tract of macaques with a predilection for the lung, infecting alveolar epithelial cells and causing diffuse alveolar damage (Fig. 3a). Furthermore, we show by semiquantitative analysis that H1N1v virus causes significantly more severe pulmonary lesions than seasonal H1N1 virus in macaques (Fig. 10). This corresponds with an earlier study in which qualitatively more severe lung lesions were observed in macaques infected with an early US isolate of H1N1v virus, A/California/04/09, than in those infected with a recent human H1N1 virus. 9 Samples for microarray analysis were obtained at 1 time point post infection only and, unfortunately, lesional lung samples could only be collected from 1 animal inoculated with seasonal H1N1 virus and 3 animals with H1N1v virus. Given this limitation, similar mRNA expression profiles between the more severely affected lungs in the H1N1v virus infection and those in the seasonal H1N1 virus infection (Fig. 12) do not suggest an intrinsically different molecular pathogenesis. Instead, the more severe pulmonary lesions are associated with more abundant virus in the lower respiratory tract by immunohistochemistry (Fig. 10).
Diffuse alveolar damage, as observed in the macaques infected with the H1N1v virus, is the pathological correlate of acute respiratory distress syndrome, 19 as diagnosed clinically in people who died with H1N1v virus infection. 15 The character of the lesions and the cell types targeted in the alveoli of the macaques is qualitatively similar to those found at autopsy of people dying from rapidly progressive pneumonia during the influenza pandemics of 1918, 1957, and 1968. 22
Interestingly, the predilection of the H1N1v virus to infect type I pneumocytes of cynomolgus macaques corresponds more to the attachment pattern of avian influenza viruses than of human influenza viruses. In virus histochemistry studies, we found that avian influenza viruses attach mainly to type I pneumocytes in the macaque lung, whereas human influenza viruses show little or no attachment at all. 23,24 A possible explanation for this apparent discrepancy is given by a recent carbohydrate microarray study. 3 In this study, 2 H1N1v viruses bound not only to the majority of α-2-6–linked sialyl sequences—“human-type” oligosaccharide receptors—but also to a considerable range of α-2-3–linked sialyl sequences—“avian-type” oligosaccharide receptors. This was in contrast to a seasonal human H1N1 virus, which bound exclusively to α-2-6–linked sialyl sequences. This suggests that H1N1v virus is able to attach both to human-type and to avian-type oligosaccharide receptors and may explain, at least in part, the higher virus replication and severity of lesions in macaque lungs than observed with seasonal H1N1 virus infection.
The H1N1v virus causes milder clinical signs and lesions in cynomolgus macaques than either avian H5N1 virus 13,20 or 1918 H1N1 virus. 11 By histopathology, this was shown mainly by less extensive edema, inflammation, and necrosis in the lung parenchyma. These differences mirror those of case fatality rates in humans from these respective infections. 2,17 Changes in the pathogenicity of the H1N1v virus as it further adapts to its new human host need to be followed closely, because adaptation may involve increased or decreased pathogenicity. Increased pathogenicity may have occurred during the 1918–1919 influenza pandemic, when a first wave of mild disease was succeeded by one with higher disease impact and mortality. 18 Decreased pathogenicity may occur if the H1N1v loses its ability to bind to α-2-3–linked sialosaccharides and therefore has less ability to infect the lower part of the respiratory tract. 3
The absence of influenza virus antigen expression in the trachea and bronchi of the H1N1v group contrasts with the results in ferrets infected with the same virus, where tracheal and bronchial epithelial cells showed abundant influenza virus antigen expression. 16 One possible explanation is differences in pattern of virus attachment between these 2 species: human influenza viruses attach more abundantly to tracheal and bronchial epithelium of ferrets than of macaques. 24 Another possible explanation is the different route of inoculation in the 2 experimental infections: intranasal in ferrets versus intratracheal in macaques.
The absence of influenza virus antigen expression in respiratory tract tissues of the seasonal H1N1 group is likely due to the lower virus titers that were present in the respiratory tract samples from animals inoculated with seasonal H1N1 virus, compared with the H1N1v virus inoculated animals (Fig. 11). The lack of virus antigen expression in tissues where virus was cultured may be due to the low quantity of viral antigen present that is below the detection limit of the immunohistochemical method. Recent pathogenesis studies in ferrets revealed comparable results in which H1N1v virus could be detected in the trachea and lung 3dpi, whereas no virus could be detected in these organs in animals that were inoculated with seasonal H1N1. 16
We show here that the H1N1v virus infects alveolar epithelial cells in a nonhuman primate model, resulting in diffuse alveolar damage that correlates with the clinical observations of acute respiratory distress syndrome in fatal human cases. Comparison with macaques infected by a seasonal human influenza virus, which is well adapted to its human host, shows that the newly emerged H1N1 virus causes more severe lung damage, which is associated with the presence of more virus in the lung parenchyma.
Footnotes
Acknowledgements
We thank T. Bestebroer, L. Leijten, M. Bijl, F. van der Panne, and P. van Run for assistance.
The authors declared that they had no conflicts of interests with respect to their authorship or the publication of this article.
This work was financed through the Virgo consortium, an Innovative Cluster approved by the Netherlands Genomics Initiative and partially funded by the Dutch Government (BSIK 03012) and through NIAID-NIH contract HHSN266200700010C.