
Abstract
The encephalitides caused by Venezuelan (VEEV), eastern (EEEV), and western (WEEV) equine encephalitis viruses are important natural diseases of horses and humans and potential agents of biowarfare or bioterrorism. No licensed vaccines or specific therapies exist to prevent or treat human infections with VEEV, EEEV, or WEEV. Well-characterized animal models are needed to support the development of such medical countermeasures under the United States Food and Drug Administration’s “Animal Rule.” This review focuses on the pathological features and pathogenetic mechanisms of these alphaviral encephalitides in animal models, with an emphasis on aerosol infections. Infection of mice, nonhuman primates, and other species with VEEV, EEEV, and WEEV causes encephalitis and often death. There is great variability in the specific manifestations of disease in the different models, however. Many aspects of the disease in animal models and in humans remain to be characterized using modern methods. Especially needed is a better understanding of the fundamental mechanisms involved in 3 key phases of the pathogenesis of alphavirus encephalitis. These are the early extraneural phase, the process of neuroinvasion itself, and virus and host factors related to neurovirulence. A greater understanding of these aspects could provide avenues for the development of medical countermeasures and better establish suitable animal models of alphavirus encephalitis for testing them under the Animal Rule.
The genus alphavirus in the family Togaviridae contains a number of arthropod-transmitted RNA viruses that are pathogens of animals and humans. The alphaviruses are generally divided into 2 groups. The old-world alphaviruses, including Chikungunya, O’nyong-nyong, and Ross River viruses, typically cause acute flulike illnesses with debilitating polyarthritis. The new-world alphaviruses, including Venezuelan equine encephalitis (VEE), eastern equine encephalitis (EEE), and western equine encephalitis (WEE) viruses, cause disease in both equids and humans that exhibit overt encephalitis in a significant percentage of cases. VEE virus (VEEV) often causes massive epizootics in horses and spillover epidemics in humans, whereas EEE virus (EEEV) and WEE (WEEV) viruses typically result in individual cases or limited outbreaks in both horses and humans. 8,45,63,84 Typical of other alphaviruses, VEEV, EEEV, and WEEV are maintained in nature by cycling between various vertebrate hosts and vector mosquitoes. Several comprehensive reviews summarizing the virological, epidemiological, and clinical aspects of the alphaviruses have been published. 8,45,63,84
Because of their high infectivity, ease of production, stability, potential for aerosolization, and consistent induction of debilitating disease, VEEV, EEEV, and WEEV are of concern as bioweapons. The susceptibility of humans to aerosol infection evident in reported cases of laboratory infections by VEEV, EEEV, and WEEV raises the level of concern. 34,82 VEEV in particular is deemed a significant biothreat 63,84 and overall is probably of greater consequence because of its ability to produce self-sustaining natural outbreaks and overt disease in nearly all human infections. 6 Not surprisingly, VEE is the most well-studied of the alphavirus encephalitides and is therefore discussed in greater detail in this review. An overriding aspect of research related to the alphaviruses is that no licensed vaccines or other specific medical countermeasures exist to protect humans against infection by VEEV, EEEV, or WEEV. Thus, research involving animal models of alphaviruses remains active. Recent studies have concentrated on the development of vaccines and on better characterizing the pathogenesis of VEE, EEE, and WEE. Understanding the viral and host factors that contribute to the development of encephalitis in particular might provide avenues for the development of therapeutics. In this light, we review many aspects of the current animal models of VEE, EEE, and WEE but note that other animal models involving encephalitic alphaviruses, such as Sindbis virus and Semliki forest virus, have also been studied. Their inclusion is beyond the scope of this review.
Venezuelan Equine Encephalitis
VEE virus actually refers to a complex of viruses, designated among 6 subtypes (I-VI; each subtype may be further categorized) that differ in terms of epidemiology and pathogenicity for humans and equids, primarily horses. Key aspects of the virology and epidemiology of VEEV have been reviewed. 8,45,63,84,87 Of major importance is the differentiation between enzootic and epizootic strains of VEEV. Enzootic strains of VEEV, designated among the subtypes ID, IE, IF, II, III, IV, V, and VI, are those that cycle naturally, mainly between Culex mosquitoes and small mammals, especially rodents. Importantly, horses do not amplify enzootic VEE viruses, and these viruses are typically considered to be avirulent for them. Horses are, however, highly susceptible to the epizootic VEEV viruses characterized as IA/B and IC subtypes. These epizootic viruses are thought to arise predominantly by genetic mutation of enzootic viruses. Morbidity and mortality rates in horses infected with epizootic viruses are high and involve a high incidence of encephalitis. 15 Not only are horses susceptible to the epizootic viruses, they amplify them greatly and the resulting viremia permits mosquito transmission and therefore fuels the epizootics. In short, epizootics of VEE require 3 key factors: the appearance of an epizootic (equine virulent) virus, a susceptible (ie, nonimmune) population of horses, and ample numbers of competent mosquitoes. Humans are susceptible to both enzootic and epizootic VEE viruses. Epidemics occur as a consequence of accidental spillover during the epizootics as humans become infected by mosquitoes that previously fed on infected horses. Person-to-person transmission of VEEV has not been reported. Human infections with enzootic strains also occur but as individual cases or small outbreaks in areas where these viruses circulate. A number of major VEE epizootics and epidemics have been documented in North, Central, and South America. The most recent significant outbreak occurred in Venezuela and Columbia in 1995, resulting in 100,000 or more human cases. 95
VEE in Humans
VEE in humans, whether due to enzootic or epizootic viruses, is typified by nonlethal, nonspecific, incapacitating illness in which fever, headache, malaise, myalgia, sore throat, and vomiting are the most common features. Lymphopenia and elevated hepatic enzymes are commonly seen during acute illness. Although nearly all human infections with VEEV are believed to be symptomatic, 24,75,84 only in a small percentage of natural cases of VEE (0.5% of adults and up to 4% of children) is central nervous system (CNS) infection apparent. 76 In such cases, CNS involvement represents a second phase of disease that usually follows the acute febrile phase by a few days. The severity of neurological disease can range from somnolence and mild confusion to seizures, ataxia, paralysis, and coma. Mortality rates in neurological cases are as high as 35% of children and 10% of adults. 6 Long-term neurological deficits, abortions, and teratogenic effects have also been reported due to VEEV. 15,68,95 In addition, a fulminant lethal form of VEE that involves a short clinical course and extensive lymphoid damage, accompanied by little or no evidence of CNS infection, has been described. 44 Another key characteristic of VEE is the susceptibility of humans to aerosolized virus. This is exemplified by the occurrence of a high number of laboratory-acquired infections involving VEEV aerosols. 34,82
The pathogenesis of VEE in humans is not well characterized, attributable in large part to a lack of data obtained during various outbreaks. Especially lacking is the availability of autopsy material from lethal cases of VEE and the application of modern immunological and molecular methods to these materials and to clinical samples. Nonetheless, available information indicates that VEE in humans shares some key features of disease in several species of animals. Biphasic febrile illness with CNS manifestations and damage to lymphoid tissues are considered the cardinal features of the more severe consequences of VEE in humans. This is based mainly on findings from 21 lethal encephalitis cases during an outbreak in the 1960s 15 and limited analysis of acute, fulminant VEE. 44
Gross findings in affected humans include cerebral edema; 44 however, the gross pathology of human cases of VEE has not been well described. The major histopathological findings in fatal cases in humans with documented infection and exhibiting clinical encephalitis were found in the brain, lymph nodes, spleen, gastrointestinal (GI) tract, liver, and lung. 15,44 Edema, congestion, and mild meningitis were the most commonly reported lesions in the brain. Meningeal infiltrates were composed of lymphocytes, mononuclear cells, and neutrophils. Inflammatory cells extended into the Virchow-Robins spaces and the surrounding neuropil in a minority of cases. Vasculitis and perivascular hemorrhage also were evident in a minority of cases. Although cerebritis was a relatively common finding in these cases, the overall distribution of histological lesions in the brain has not been thoroughly documented. Neuronal changes have not been described, to our knowledge. The main histological changes in lymph nodes, spleen, and GI lymphoid tissue (GALT) were lymphocyte degeneration, lymphoid depletion, and follicular necrosis, accompanied by infiltrates of neutrophils. Lung lesions were present in nearly all cases, and liver lesions were found in a majority of cases. Moderate to severe, interstitial pneumonia with infiltration of alveolar septa by neutrophils, lymphocytes, and macrophages was the key finding in the lungs. In the liver, hepatocellular degeneration and necrosis and minimal inflammatory infiltrates were the main findings. Vasculitis in lymphoid and other tissues was also considered a key finding in these cases. The pathological analysis of reported human cases did not include the use of immunohistochemistry or electron microscopy.
Animal Models of VEE
Experimental animal studies with VEEV have primarily focused on attempts to characterize the early pathogenesis of VEE, the route of neuroinvasion, and host and viral factors that contribute to the development of encephalitis. Key to interpreting these studies is an understanding that a variety of VEEV strains or genetically engineered mutants have been used. Available variants of VEEV involve virulent strains obtained from natural infections, serially passaged isolates, and genetic mutants manufactured by design. Of particular note is the availability of a full-length complementary DNA clone of the virulent Trinidad donkey strain of VEEV (designated as V3000), 14 from which a variety of genetically modified VEE viruses have been generated and characterized. These mutant viruses have been used to understand key viral determinants in the pathogenesis of VEE, and some have been studied as potential vaccines, because of their various degrees of attenuation.
Several species of animals have been used as models of VEE. 26,42,45,47,63,84,89 Horses and other equids are highly susceptible to epizootic strains of VEEV, with morbidity in epizootics estimated at 40%–60% of susceptible horses and mortality rates of around 50%. 18,26,58,69,84,94 Clinically, these species develop fever, depression, weakness, circling, and convulsions, along with leukopenia, lymphopenia, and decreased hematocrit. Pathologically, VEEV infection of equids results in meningoencephalitis characterized by infiltrates of lymphocytes, mononuclear cells, and neutrophils in the meninges, surrounding vessels, and the neuropil. Gliosis, satellitosis, some degree of neuronal damage, hemorrhage, and vasculitis are additional features of the brain lesions in equines. The cerebral cortex is most severely affected, but most parts of the brain exhibit some histological changes. The severity and nature of the changes in the brain vary among individual cases and also depend on the time course of infection and other factors. In addition to CNS changes, lymphoid necrosis is a common feature of VEE in equids. Other reported lesions include hepatic and adrenal necrosis, as well as lesions in the bone marrow, heart, pancreas, and kidneys. The pathogenicity of endemic strains of VEE in experimentally inoculated equids is variable. 18,74 Although horses are clearly important with respect to VEE as a natural disease, for reasons owing mainly to their size, they are limited in their use as an experimental model of VEE.
Among the laboratory animals investigated so far, hamsters, rabbits, and guinea pigs develop an acute, fulminant disease typified by massive necrosis of lymphoid tissues, in particular the GALT. 27,42,43,93 These animals usually die before the onset of CNS disease, so they are also of limited use as models of VEE in humans. Mice and nonhuman primates (NHPs) are the 2 animal models considered most relevant to human VEE. Therefore, we review available information about these 2 models in some detail.
Pathology of the Mouse Model of VEE
A number of important aspects of VEEV infection have been studied in the mouse model, including ones relevant to the natural course of infection and also relevant to biological warfare (ie, aerosol infection). The virulence of a variety of strains of VEE has been tested in mice. 12,29,53,83,92 The median lethal dose (LD50) of virulent epizootic strains of VEEV in mice ranges from 1 to 30 plaque-forming units (pfu), depending on the route of inoculation and the strain and age of the mice. 53,63,86 Immunocompetent mice typically succumb to VEE within 6–9 days of infection.
Natural (mosquito-transmitted) infection, modeled experimentally by subcutaneous (SC) or footpad inoculation of mice with virulent strains of VEEV, results first in infection of dermal dendritic cells (DCs). The DCs carry the virus to the draining lymph nodes, 54 possibly supplemented by movement of labile virus through the draining lymphatics. In the lymph node, VEEV first begins to replicate by about 4 hours postinfection (PI). 13,29 VEEV enters the bloodstream as early as 12 hours PI, reaches serum titers of approximately 1 × 106 pfu/mL, and subsequently infects a variety of additional tissues. In particular, VEEV is strongly tropic for lymphoid tissues in mice, with infection being evident in the spleen, GALT and nasal-associated lymphoid tissues, thymus, bone marrow, and nondraining lymph nodes. Virus replication occurs especially within mononuclear phagocytes in these locations. 30,90 Active infection also occurs in exocrine pancreatic cells, hepatocytes, the chemosensory epithelium of the vomeronasal organs, Bowman’s glands, and the odontogenic and ameloblastic epithelia and dental pulp of the teeth. 42,83,90
Mice effectively clear VEEV from lymphoid tissues, the blood, and other tissues by 3–4 days PI, but this is of little consequence given that infection of the brain is already evident by 36–48 hours PI after inoculation of the skin. 10 Neuroinvasion involves appearance of VEEV first within the olfactory bulbs, 10,29,63,72,73,83,90 not unlike other arboviral encephalitides. Importantly, virus particles in the blood appear to escape through fenestrated capillaries beneath the olfactory mucosa of the nasal tract, followed by invasion of nearby unmyelinated axons of the olfactory nerves. 10 Virus then passes into the olfactory bulbs of the brain via the olfactory nerves. Direct invasion of the brain via the bloodstream, although suggested, 73 does not appear to be a significant characteristic of VEEV in mice. A second conduit for neuroinvasion, however, is via the trigeminal nerves subsequent to virus infection of the teeth. 10,83
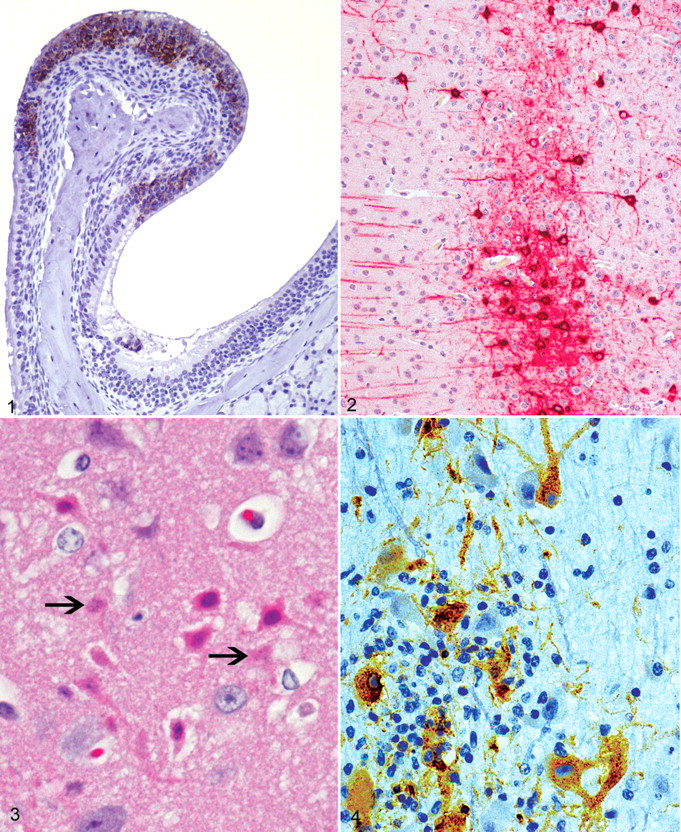
Infection of the brain following aerosol inoculation takes a more direct route. A key factor in this regard is the susceptibility of olfactory neurons to infection by VEEV, as they incur overwhelming infection via aerosols (Fig. 1 ). 73,83,90 This tropism for olfactory neuroepithelium is specific, as neither the respiratory nor squamous epithelia of the nasal tracts are infected by VEEV. 63 However, sustentacular cells and basal cells of the olfactory lining do show evidence of infection. It is noteworthy that the bipolar olfactory neurons contact inspired air via their superficial processes and also synapse directly with neurons in the olfactory bulbs via their axons, which form the olfactory nerves (fila olfactoria). By targeting olfactory neurons, VEEV is thus provided a direct and rapid conduit to the brain.
By both the dermal and aerosol routes of inoculation, therefore, neuroinvasion by VEEV in mice mainly involves the olfactory system. Accordingly, 2 particular factors are worth noting. First, aerosol infection results in more rapid neuroinvasion, occurring as early as 16 hours PI. 73 This point is potentially important in regard to the effectiveness of early antiviral host factors. Second, neuroinvasion by aerosol infection does not require the development of viremia. This point is of particular consequence because serum neutralizing immunoglobulin (Ig)G antibodies that afford protection from peripheral infection may not protect against aerosol infection. In fact, protection against aerosol infection appears to rely instead on the induction of mucosal IgA antibodies, 35,36 a factor that has complicated the development of vaccines effective against both natural and aerosol infection by VEEV.
Once in the olfactory bulbs, VEEV spreads rapidly to the brain proper, appearing initially in the lateral olfactory tracts, the pyriform cortex, and the olfactory tubercles. This is followed by the appearance of virus in more caudal locations, including the basal nuclei, thalamus, neocortex (Fig. 2 ), midbrain, caudal brainstem, and cerebellum. How efficiently and rapidly VEEV reaches particular regions of the brain depends on the route of administration (eg, generally faster with aerosol infection) and on the strain of virus used for infection. The thalamus, given its role as a relay station, may be important in the rapid transport of VEEV to multiple locations in the brain and subsequently the spinal cord. Neurons are overwhelmingly the main target of infection in the brain and spinal cord. 42,83,90 This includes a variety of subtypes of neurons such as pyramidal neurons of the neocortex, granular neurons of the hippocampus, Purkinje and granular neurons in the cerebellum, motor neurons in the caudal brainstem, and anterior horn cells in the spinal cord. Virus antigens and mature virions are evident in the cell bodies of neurons and also in axons. Macrophages in the brain also show evidence of infection. 90 Astrocytes are susceptible to VEEV in vitro 78 but seem not to be strong targets of infection in vivo. 63 Additional resident CNS cells likewise demonstrate little if any infection. In particular, endothelial cells, the choroid plexus, and ependymal cells are not considered targets of VEEV. Despite the fact that substantial host immune and inflammatory alterations result from CNS infection in mice, as we review subsequently, virus infection of neurons in the brain and spinal cord appears sufficient to explain the majority of clinical signs evident in mice and to account for the uniform lethality of virulent strains of VEEV.
Mice experimentally inoculated with attenuated strains of VEEV exhibit a range of clinical signs, tissue tropism, neuroinvasiveness, and lethality. 29,53,83,85 Importantly, the outcome of infection by a particular strain of virus depends to a great extent on the route of inoculation. For example, the modified live vaccine virus TC-83 given by dermal inoculation of C3H/HeN and BALB/c mice is essentially avirulent, 35 yet aerosol inoculation of both strains of mice with TC-83 virus results in uniform infection of the brain and 100% lethality in the C3H/HeN mice. 83 Similarly, the genetically engineered strain V3014 is neither neuroinvasive nor lethal in CD-1 mice inoculated in the footpad, but intracerebral (IC) administration of this virus produces 100% mortality. 29 The engineered strain V3526 is not neuroinvasive following dermal inoculation and is consistently neuroinvasive following aerosol administration yet has only limited neurovirulence and is nonlethal following both aerosol and direct IC inoculation. 53,83,84 The combined information provided by these and other studies with VEEV indicates that the processes of neuroinvasion and neurovirulence are at least somewhat independent of each other and that the outcome of infection in mice with different VEE viruses involves a variety of viral and host factors.
Mice infected with VEEV demonstrate lethargy, huddling, dehydration, weight loss, tremors, and paralysis or paresis. 85 Seizures are rare in mice with VEE. 25 Histological lesions are present in both neural and extraneural tissues. The CNS lesions are characterized as necrotizing panencephalitis and myelitis in their fully developed forms. They develop in regions of the brain according to the appearance of the virus in those locations; for example, lesions are seen initially in the olfactory bulbs and later in more caudal structures. In general, lesions become evident histologically or ultrastructurally about 24 hours after virus is present in a particular location. 73,83,85,90 Early changes involve the outer portion of the olfactory bulbs and are characterized by congestion and minor hemorrhage, damaged endothelial cells, perivascular edema, minimal necrosis, and infiltration of a few neutrophils and mononuclear cells into the meninges and perivascular regions. Similar lesions extend into more caudal locations at later times and are accompanied by increased amounts of necrotic cellular debris, thicker perivascular cuffs composed increasingly of mononuclear cells, rarefaction of the neuropil, and infiltration of the neuropil by neutrophils, lymphocytes, and macrophages. One feature that is notably lacking in mice infected with VEEV is outright vasculitis.
Not surprisingly, neuronal damage is a key component of the brain lesions of experimental VEE in mice. The type of neuronal damage in VEE has been ascribed to both necrosis and apoptosis. 41,77,83,85 In our experience, the morphological changes that occur in mouse neurons during the course of VEE depend on the type of neuron involved. 85 Histologically, larger neurons show morphologic changes typical of necrosis (Fig. 3 ). This is particularly evident in the pyramidal layer of the pyriform cortex. Conversely, granule-type neurons, including those of the hippocampus and the cerebellum, show morphological changes more typical of apoptosis. The mechanism of neuronal damage in different regions of the brains of VEEV-infected mice, however, has not been well studied. This is in contrast to the prototype alphavirus Sindbis, which has been more extensively studied in this regard and shown to cause neuronal apoptosis in an age-related manner that involves regulation by cellular bcl-2. 31,32,49 In addition to neuronal damage, damage to glial cells and abundant lymphocytolysis also occur in the brains of mice with VEE. Astrocytosis is reported to occur both in areas where active infection is evident as well as areas where the virus appears not to be present. 77
Immunopathological and other host factors appear to contribute in significant ways to the pathogenesis of encephalitis in VEE. For example, the survival times of mice following infection with VEEV are longer in SCID mice, T-cell depleted mice, and mice treated with antithymocyte serum than in immunocompetent mice. 9,96 Furthermore, the increased survival time in immunocompromised mice occurs despite the presence of high titers of virus in the brain. These findings have been attributed mostly to decreased numbers of inflammatory cells, in particular CD8+ T lymphocytes, in the brains of the immunocompromised mice. Other studies have pointed to additional cellular and soluble factors that may contribute to the severity of encephalitis. One study attributed neuronal damage in parts of the brain without apparent VEEV infection to activation of astrocytes. 77 The same study also reported upregulation of a variety of apoptosis-signaling molecules in the brains of infected mice, although the cellular source of these mediators was not determined. Additional VEE studies have shown upregulation of a variety of proinflammatory mediators in the brains of VEEV-infected mice, such as tumor necrosis factor-α, inducible nitric oxide synthase, interferon-γ, interleukin-1, interleukin-6, chemokines, and Toll-like receptors. 28,77 –81 Combined, these reports support the notion that various host factors contribute to the severity of encephalitis in VEE.
Severe damage to lymphoid tissues is another prominent feature of VEE in mice. 9,42,83,90 Early in the course of disease, lymphoid damage becomes evident as diffuse necrosis of the outer cortex of follicles in lymph nodes draining the site of inoculation. Infiltrates of neutrophils are often evident in areas of lymphoid necrosis. There is variable necrosis in the remaining lymph nodes and the mucosa-associated lymphoid tissues. In the spleen, necrosis affects much of the white pulp but may be most severe in B-cell areas. Despite the extensive lymphoid necrosis evident early in VEE, lymphoid regeneration occurs rapidly in immunocompetent mice, and marked lymphoid hyperplasia is present during the latter stage of disease.
Lesions are also evident in a variety of other tissues in which active VEEV infection occurs in mice. These lesions typically involve some degree of necrotic change and usually some measure of inflammation, depending on the timing of infection and the particular tissue involved. One prominent lesion worth mentioning is the necrotizing rhinitis that typically occurs within 1–2 days of exposure of the olfactory mucosa to infectious virus. This and other lesions in a variety of tissues seem of little consequence, however, compared with those that occur in lymphoid tissues and the CNS.
Pathology of the Nonhuman Primate Model of VEE
Most VEE studies involving NHPs have used the cynomolgus macaque (Macaca fascicularis), although a few used rhesus macaques (Macaca mulatta). 11,26,55,56,63,67,88,89 Both species are susceptible to enzootic and epizootic strains of VEEV by various routes, including aerosols. With the exception of direct IC inoculation, most macaques infected with VEE viruses survive infection. Key features of VEE in macaques include the development of fever, viremia, and lymphopenia within 1–3 days of infection and signs of encephalitis a few days later in some cases. The numbers of animals that developed viremia, fever, and/or neurological signs varies among reported studies, probably because the investigators used different combinations of virus strain, route of administration, and species of macaque. Only a few studies have documented the pathological findings in NHPs infected with VEEV. 11,26,56,89 They are somewhat limited in their value, though, because ancillary methods such as immunohistochemistry, in situ hybridization, and electron microscopy were not available or were not included. Also, the viruses used were not characterized according to currently accepted means for VEEV. Nevertheless, these studies have provided the best available descriptions of the pathology of VEEV in NHPs.
In the most thoroughly characterized NHP study, 26 67 rhesus macaques were infected with various doses of an equine-virulent strain of VEEV by the intraperitoneal (IP) route and were euthanatized at multiple time points. Similar to humans with VEE, the infected macaques developed transient viremia around 1–2 days PI and a biphasic fever with the initial phase at 12–72 hours PI and the second phase beginning around day 5. Otherwise, the macaques exhibited no evidence of clinical disease. Histological findings predominantly affected lymphoid tissues and the brain. Lesions in the brain were consistent findings, evident in 18 of the 20 macaques euthanatized between 14 and 21 days PI and also in some animals euthanatized earlier. Lesions typically developed around day 6 PI in the olfactory cortex, thalamus, and hypothalamus and spread throughout the brain by day 8. The most characteristic brain lesions were multifocal perivascular cuffs, composed mainly of lymphocytes, and gliosis. These changes were most severe from days 14 to 21 PI but reportedly varied in severity from animal to animal and also varied among different regions of the brain for individual macaques. In particular, the thalamus was the site of the most intense inflammation, whereas the occipital cortex, cerebellar cortex, and hippocampus were generally spared. Lesions in the spinal cord were present but were not severe. Neuronal damage associated with glial nodules was evident but was uncommon in this study. Additional features of encephalitis that were notably lacking in these macaques included hemorrhage, vasculitis, meningitis, demyelination, and the presence of neutrophils in foci of inflammation. Most lesions resolved after 5 weeks or more, with few residual histopathological changes at 12–13 weeks PI. In the lymph nodes and spleens of these macaques, lymphocyte destruction was evident by day 2 PI. Lymphoid necrosis was most intense in the follicles. Removal of cell debris occurred between days 2 and 4 PI, and this was followed by lymphoid hyperplasia. Focal hepatic and myocardial necrosis and inflammation attributable to virus infection were rare findings in these macaques. However, this study did not provide direct evidence of virus infection of tissues associated with such lesions by means of immunohistochemistry or other methods, so it is somewhat difficult to differentiate VEE-associated lesions from incidental lesions in some instances.
Other studies involving pathological analysis of NHPs with VEE reported similar findings. The most notable difference was in macaques infected by the intranasal (IN) route, in which lesions were evident in the olfactory bulbs at 48 hours PI, 11 compared with about 6 days PI for macaques infected by the IP route. Neuronal damage with neuronophagia and infiltration of the brain by neutrophils were additional features of these macaques. This report seemed to imply that the degree of neuronal damage was more severe in the macaques infected IN than those from other studies involving alternative routes of inoculation.
Recent studies of VEE involving macaques performed at our institute have been limited. Cynomolgus macaques infected with IE and IIIA strains of VEEV by aerosol developed fever, viremia, lymphopenia, and clinical signs of encephalitis but recovered. 66,67 These studies did not use pathological analysis. In separate studies, cynomolgus macaques infected with an IA/B strain of VEEV intranasally developed viremia and mild clinical disease. 56 Two macaques euthanatized at 21 days PI and examined histologically demonstrated multifocal nonsuppurative encephalitis with gliosis, similar to the nonhuman primate studies already discussed. In this same study, macaques infected with the same virus IC developed fulminant lethal encephalitis. In these cases, moderate to severe inflammation and gliosis were evident in the brain, but the lesions were not otherwise described.
A serial sacrifice study recently conducted at our institute focused on collection of medical diagnostic samples before and after aerosol exposure of NHPs to VEEV. Eight cynomolgus macaques were administered aerosols of the virulent Trinidad strain of VEEV and 2 macaques each were sacrificed on days 1, 2, 3, and 4 PI. Some aspects of this study have previously been published although they did not include pathological findings. 33,48,59 We reviewed archived pathology samples from this study and noted immunohistochemical evidence of infection in mononuclear cells of the axillary and mesenteric lymph nodes and the spleens of all but 2 cases (one on day 1 and one on day 2 PI; N. Twenhafel, unpublished). Evidence of infection of the brain, however, was limited to a single macaque sacrificed at day 4 PI. This case exhibited a few to moderate numbers of cells with VEE virus antigen, primarily individual neurons or small foci of immunolabeled cells in the region of the olfactory tubercle and nearby anterior commissure (Fig. 4). Infected cells were accompanied by mild to moderate nonsuppurative encephalitis characterized by the presence of perivascular cuffs, gliosis, satellitosis, neuronal death, and a few microhemorrhages. Otherwise, evidence of active virus infection of tissues in these cases was lacking, despite gross and histopathological analysis of a full complement of tissues, including rostral brain structures, cerebrum, cerebellum, brain stem, pituitary gland, eyes, nasal septum, cranial nerves I, II, III, V, and teeth. This study did not include macaques sacrificed at later times when brain lesions typical of VEE are expected to reach their fullest expression.
The specific viral and host mechanisms that drive the processes of neuroinvasion and neurovirulence remain 2 of the major gaps in our knowledge of the pathogenesis of VEE. Despite the limitations of the various NHP studies, they are informative in that they collectively show that neuroinvasion and neurovirulence both occur in a limited fashion in NHPs inoculated with VEEV by routes other than IC. In these respects, the NHP model of VEE appears more similar to human cases of VEE than does the mouse model, in which equivalent VEE viruses are more frequently neuroinvasive and neurovirulent. The reasons for these apparent differences are not known, although anatomical differences in the olfactory tract of mice compared with NHPs may be partially explanatory. It is difficult to speculate further about these differences, however, because the NHP model of VEE has not been characterized to the level of detail of the mouse model. Therefore, future NHP studies in particular should include a full and systematic characterization of the development of CNS lesions throughout the course of disease using currently available pathological methods. Using NHPs of varying age might be informative as well. The use of additional NHP species should be explored further, as suggested by recent alphavirus studies using marmosets and owl monkeys 1,21 and the growing utility of African green monkeys as models of human diseases like leishmaniasis, anthrax, and tularemia.
Eastern Equine Encephalitis
Various strains of EEEV have been isolated from humans, horses, birds, and small mammals in eastern regions of the Americas from Canada to Argentina. 8 Most human infections with EEEV are asymptomatic, but when CNS involvement occurs, it results in more frequent and severe neurological signs, lesions, and sequelae than are seen with the other arboviral encephalitides. 4,17,22,84 Estimates of the mortality rate in human cases of EEE vary from 36% to as high as 75%. Similar to VEEV, the young are at greater risk of clinical disease due to EEEV. The early clinical illness involves a flulike prodromal stage of about 5 days dominated by high fever and headache. 8,17,84 Children commonly exhibit either generalized or mainly facial edema. Paresis, paralysis, respiratory impairment, altered mental state, and seizures are common neurological manifestations. Many of these signs persist for long periods in patients who survive the acute illness. In fatal cases of EEE, gross lesions in the brain include edema, meningeal congestion, hemorrhage, and malacia. 17 Histopathological findings involve neuronal damage; meningeal, perivascular, and parenchymal infiltrates of neutrophils and mononuclear cells; gliosis; demyelination; vasculitis; and thrombosis. 8,16,17,37 Lesions are generally widespread in the brain, although the basal nuclei, thalamus, and brainstem are particularly targeted.
In general, EEEV has not been as well studied in animals as VEEV. Comparatively few of the published studies have been conducted recently and have used state-of-the-art methods. Nonetheless, a variety of animal models of EEE have been described, including mice, rats, hamsters, guinea pigs, rabbits, and NHPs. 19,20,39,40,51,62 –64,91 Here, we focus on the mouse, guinea pig, hamster, and NHP models, which are the best studied and have been shown to manifest features of CNS infection most applicable to our understanding of the human condition.
Pathology of the Mouse Model of EEE
Mice infected with EEEV demonstrate both similarities and differences compared with human cases of EEE as well as the mouse model of VEE. As is the case generally with alphaviruses, the resistance of mice to infection by EEEV increases with age. Similar to VEEV, mice are susceptible to infection with EEEV by cutaneous, IN, and IC inoculation and by aerosol administration. Calculated LD50 values for EEEV in mice vary from less than 1 pfu by IC inoculation to 400 pfu by the IP route, 500 pfu by aerosol, and 1250 pfu by SC administration. 7,63 Also similar to VEE in mice and mimicking at least some aspects of EEE in humans, young mice infected with EEEV develop a biphasic disease course manifested by initial virus replication in peripheral tissues followed by viremia, CNS invasion, and encephalitis. Infected mice often exhibit seizures but not paralysis. 25 This point is in contrast to mice with VEE, which display paralysis but rarely have seizures. Mice typically die between days 4 and 6 PI after peripheral inoculation with EEEV. 25,91
Also in contrast to VEE, EEEV does not replicate well in mouse lymphoid tissues in vivo. In vitro, EEEV fails to replicate specifically in macrophages and DCs. 25 It appears that EEEV can bind to, enter, and uncoat in these cells, but then replication arrests because of failure to translate the subgenomic virus message. Similar phenomena may occur in human cells. For example, VEEV infects and replicates efficiently in human leukocytes in vitro, whereas EEEV replication is restricted in these cells. 50 In mice, primary EEEV replication in vivo occurs instead in fibroblasts, skeletal muscle, and osteoblasts. 91 This ability of EEEV to replicate in mesenchymal cells, abundant and active in growing mice, has been proposed as one reason for the greater susceptibility of young mice to EEE. In addition, the inability of EEEV to replicate in lymphoid tissues may help explain the fact that clinical signs of EEE arise later than they do in mice with VEE. 25
Infection of mice with virulent strains of EEEV results in neuroinvasion, widespread infection of neurons, and severe encephalitis. Vasculitis, considered an important pathological feature of encephalitis in human cases of EEE, is not typical in mice, however. 62,63 Compared with VEE in mice, neuroinvasion by EEEV reportedly appears to involve entry into the brain directly from the bloodstream, rather than through olfactory or other peripheral nerves. 91 After SC inoculation, EEEV first appears in neurons of the brain as early as 24 hours PI using immunohistochemistry and in situ hybridization. The appearance of virus in multiple regions of the brain at the same time suggests entry from the vasculature. By day 4 PI, virus is present in neurons and occasional glial cells of most parts of the brain. The caudate nucleus/putamen, thalamus, and pons appear to be the most strongly targeted regions in the brain. Infection of the spinal cord has not been well studied in EEE. The major histological change in the brains of mice with EEE is widespread neuronal necrosis, accompanied by rarefaction of the adjacent neuropil in the gray matter and some white matter tracts. Mild inflammation involving infiltrates of neutrophils and some eosinophils is also characteristic.
Additional targets of EEEV outside of the brain include fibroblasts, skeletal muscle and cardiac myocytes, osteoblasts, ovarian stromal cells, keratinocytes, sebaceous gland epithelium, odontoblasts and ameloblasts in the teeth, retinal ganglion cells in the eyes, and a few olfactory neuroepithelial cells. An important extraneural lesion in young mice is abnormal bone formation characterized by loss of osteoblasts, reduced osteoid production, and cartilage hypertrophy. 91 Widespread lymphocyte apoptosis affects the thymus and secondary lymphoid tissues of these mice but is unassociated with the presence of active viral replication and thus likely represents bystander lymphocyte destruction.
We recently reported that mice were susceptible to aerosol infection by North American and South American strains of EEEV, 70 but we did not describe the pathological effects in mice as part of that report. Separate studies at our institute, however, indicate that mice infected with EEEV by aerosol show neuronal tropism and brain lesions similar to mice infected peripherally (M. Teehee, unpublished). For example, brain infection in aerosol-inoculated mice is evident at 24 hours PI, similar to peripheral inoculation. However, virus in the aerosol-inoculated mice was only evident in the olfactory bulbs of the brain at 24 hours. This finding was accompanied by evidence of infection in the olfactory neuroepithelium, suggesting that neuroinvasion following aerosol infection occurs via the olfactory tract, which is in contrast to mice peripherally inoculated with EEEV 91 but similar to mice with aerosolized VEEV.
Pathology of the Guinea Pig and Hamster Models of EEE
We recently reported results of a study in which guinea pigs were infected with EEE viruses by aerosol. 70 In this study, some animals developed clinical signs within 24 hours of infection, evident as decreased activity and dorsal tremors. These signs rapidly progressed to circling, recumbency, coma, and death. Pathologically, the guinea pigs demonstrated changes similar to mice with VEE, including virus infection of the olfactory neuroepithelium and appearance of virus in the olfactory bulbs by 24 hours PI, followed by spread to all parts of the brain by day 4 PI. Neurons were the main viral target of infection in the brain. Brain lesions were dominated by neuronal necrosis and the presence of perivascular cuffs and infiltrates in the neuropil composed of macrophages and heterophils. Vasculitis was also evident in some late-stage cases, manifest by fragmentation of the walls of vessels, intramural infiltrates of inflammatory cells, and fibrinoid necrosis. Despite the administration of virus by aerosol, only minimal amounts of virus were detected in the lungs of a few animals. Productive infection was not detected in the conducting airways, heart, liver, pancreas, kidneys, reproductive organs, thymus, spleen, mesenteric lymph nodes, bone marrow, and the GI tract. Osteoblasts were only rarely infected, in contrast to the study of EEEV in mice reviewed earlier. 91
Hamsters develop a biphasic illness due to infection with virulent EEE viruses. 20,62,63 Hamsters inoculated peripherally develop an early visceral phase, accompanied by viremia and followed by neuroinvasion and ultimately death due to encephalitis around days 4–6 PI. They exhibit fever within 24 hours of infection, vomiting and respiratory signs, and head-pressing, stupor, and coma. 62 Early EEEV targets in the hamster are the heart, liver, lungs, kidneys, lymphoid tissues, and skeletal muscle. Targeting of osteoblasts in the hamster has not been reported in the published studies. Virus is cleared from the blood and peripheral tissues, but not the brain, upon development of neutralizing antibodies. EEEV enters the brain about day 2 PI and replicates progressively thereafter, reaching titers well exceeding those in other tissues. 20 The basal nuclei, hippocampus, and brainstem are particularly targeted, although the cerebral cortex and cerebellum are also affected. Other histopathological features of EEE in the brains of hamsters include neuronal tropism and destruction, inflammatory cell infiltration of the neuropil, gliosis, and microhemorrhages. Vasculitis was reported among the features of brain lesions in the hamster in one reported study. This was based on infiltration of vessel walls by neutrophils and mononuclear cells. 62 Other features typical of vasculitis (eg, fibrinoid necrosis, leukocytoclasia) were not reported in this study.
Pathology of the Nonhuman Primate Model of EEE
EEE pathogenesis or natural history experiments characterizing the disease in various NHP species and describing their potential usefulness as animal models are limited. A study conducted in the 1960s harvested virus from a fatal human case and injected the unpassaged virus into the thalamus of juvenile rhesus macaques. 61 All animals subsequently developed acute encephalitis and were euthanatized 4 days after inoculation. Histopathologic changes included severe encephalitis with neuronal necrosis and massive loss of neurons, primarily in the forebrain. Inflammation in these areas, considered mild compared with the overwhelming neuronal destruction, consisted of pervascular cuffs and leptomeningitis.
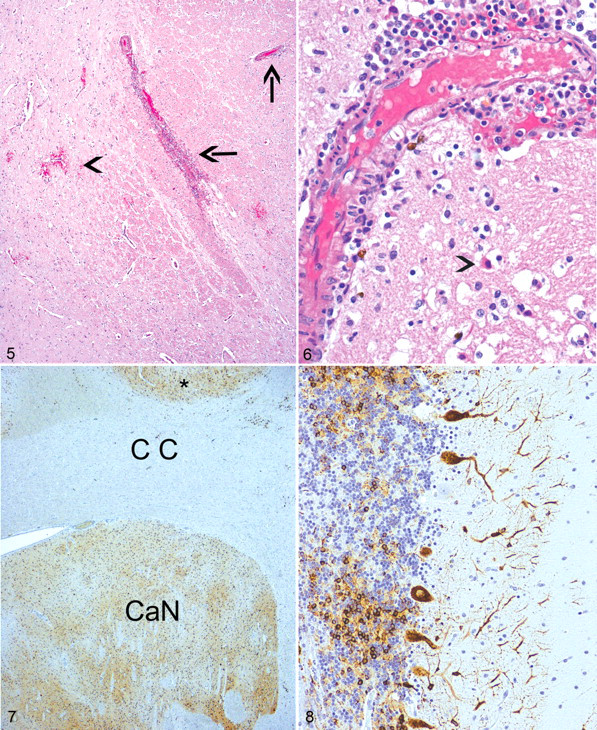
The majority of work with EEEV at our institute has focused on vaccine trials. However, a recent natural history study was conducted in which 12 adult cynomolgus macaques were challenged with aerosolized virulent EEEV. 64 Eight of the 12 macaques developed fever, elevated white blood cell counts, elevated liver enzymes, and neurologic signs and succumbed 5–9 days after challenge. Viremia was transient in some cases and undetectable in others. We examined the pathology reports from this study and reexamined archived pathology materials to assess the key pathological features of EEE in macaques. Gross necropsy results were unremarkable in all animals. The major histopathological features of animals that succumbed to disease are summarized in Table 1 . The major CNS changes were characterized as severe meningoencephalomyelitis. Specific histological features included widespread neuronal necrosis and the presence of perivascular cuffs, cellular debris, gliosis, satellitosis, edema, and hemorrhage (Fig. 5 ). Vasculitis affected cerebral blood vessels (Fig. 6 ). Microthrombi were evident rarely. The Virchow-Robbins spaces were often expanded by edema and occasionally hemorrhage. EEEV antigen was present in multiple regions of the brain (Fig. 7 ), especially in neurons (Fig. 8 ).
Key Features of Eastern Equine Encephalitis (EEE) in Nonhuman Primates Inoculated by Aerosolized EEE Virus a
IHC, immunohistochemistry; NA, not acquired at necropsy; PI, postinfection; +, mild; ++, moderate; +++, severe/marked
a Information in this table represents pathology data from cases of a previously published study. 64
In a recently published study, adult marmosets were inoculated IN with either a virulent North American (NA) or an attenuated South American (SA) strain of EEEV. 1 The marmosets inoculated with the SA virus did not develop clinical disease. Those inoculated with the NA strain developed meningoencephalitis with perivascular hemorrhage affecting the cerebral cortex but did not develop detectable viremia. Another recent study described a group of owl monkeys (Aotus nancymaae) inoculated IN and SC with virulent EEEV. 21 None of the animals displayed clinical signs of disease and none of the IN-inoculated animals became viremic. The SQ-inoculated animals manifested a viremia lasting 3.2 days on average. Two animals from each group were euthanatized on day 6 PI but exhibited no significant gross or histologic findings. Taken together, these studies suggest that NHP models develop key features of EEE in humans such as neuronal tropism and necrosis, meningoencephalitis, and vascular damage.
Western Equine Encephalitis
Strains of WEEV have been isolated from humans, horses, wild birds, and mammals from western Canada to Argentina. 8 Natural human cases of WEE generally have more features in common with EEE than with VEE. For unknown reasons, the incidence of WEE in humans and in horses has declined dramatically since the middle of the 20th century. 8,23 Accordingly, the majority of clinical and pathological studies of WEE in humans were published many years ago. On the basis of these reports, the incidence and severity of neurological signs and the mortality rate in human cases of WEE vary depending on age and probably also on virus strain, dose, and route of infection. 8,34,71,84 As with VEE and EEE, natural human cases of WEE typically show an early, flulike illness with fever, malaise, and headache. Similar to EEE, WEE results in CNS signs in a significant proportion of cases. These signs include somnolence, seizures, coma, and motor neuron dysfunction. An estimated 90% of humans with WEE under the age of 1 year show severe CNS signs. 8 The mortality rate in human WEE depends on age and other factors. Estimates of the case fatality rate of WEE range from 3% to 15%. In patients who survive infection, neurological sequelae may linger for months to years or may even be permanent. Laboratory accidents involving aerosol exposure to WEEV have been documented. The mortality rate of a few cases of aerosolized WEE in humans was 40%. 34 Histopathologically, the brains of humans who have WEE have perivascular cuffs of lymphocytes and neutrophils, multifocal necrosis, and gliosis. 3,71 Both gray and white matter are affected in multiple regions of the brain, with the basal nuclei, thalamus, and brain stem often the most severely affected. Lesions are also seen in the spinal cord.
Like EEE, WEE has not been extensively studied in animals. A few recent studies involving mice, hamsters, and cynomolgus macaques, however, offer new insights based on modern scientific methods. Older studies also investigated rats, gerbils, guinea pigs, and rabbits as models of WEE. 38,39,63
Pathology of the Mouse Model of WEE
Several studies have reported the susceptibility of mice to WEE viruses and documented differing levels of virulence according to virus strain, the age and strain of mice infected, and the route of inoculation. 2,5,52,57,60,63 The descriptions of pathological changes in these reports, however, are limited. One recent study demonstrated the virulence parameters of multiple WEEV strains administered to 6- to 8-week-old CD-1 mice. 52 The most lethal strain tested, MacMillan (MCM), was 100% fatal when inoculated IC, SC, or by aerosol and was 90% lethal by the intravenous (IV) and IN routes. The mean survival time with this strain varied from 1.9 days (IC) to 6.1 days (IV). Inoculation of the MCM strain SC produced high titers of virus in the brain and draining popliteal lymph nodes and lower amounts of virus in other tissues. Histological lesions occurred in the brains of all mice infected with the MCM strain and were characterized by laminar or multifocal neuronal necrosis, edema, mild infiltrates of lymphocytes in the meninges and subependymal areas, and occasional fibrin thrombi. Other strains of WEEV demonstrated lesser virulence than MCM and differing amounts of virus in the brain, depending on the route of inoculation. Lesions were also seen in the brains of some mice infected with the least virulent strain (IMP 181). These mice had limited neuronal necrosis and, unlike the mice given MCM, exhibited glial nodules and perivascular cuffs. Likely, these additional changes developed because the mice infected with the IMP 181 strain survived longer.
An older study with a strain of WEEV described as B-629 showed that 3- to 4-week-old mice developed widespread meningoencephalitis with necrosis, perivascular lymphoid cuffs, microglial nodules, and astrocytosis. 2 Lesions were also present in the lungs, liver, and heart of these older mice. Infection of the heart and myocarditis has been described in adult mice infected with a different strain of WEEV. 57 An interesting finding in the study using WEEV B-629 is that suckling mice infected SC developed rapidly lethal disease and died within 2 days of inoculation. They exhibited acute inflammation and/or necrosis in bone marrow, skeletal muscle, cartilage, peripheral nerves, and other mesodermal tissues but not in the brain. The results of this study are reminiscent of those seen in some species of animal infected with virulent VEE viruses, in which rapid death due to overwhelming peripheral (lymphoid) infection preempted neuroinvasion and the development of brain lesions. 27,42,43,93
Pathology of the Hamster Model of WEE
Adult hamsters infected with an unspecified virulent strain of WEEV by a variety of routes showed lethal disease characterized by tachypnea, conjunctivitis, incoordination, and seizures. 98 Infection by various routes also resulted in high titers of virus in the brain and encephalitis. The major brain lesions were neuronal necrosis, infiltration of the meninges and Virchow-Robins space by lymphocytes, astrocytosis, microgliosis, hemorrhage, and “spongy degeneration.” All parts of the brain were affected, especially the olfactory regions. A recent study using the VR-70 (California) WEEV strain also showed lethal infection of adult hamsters inoculated IP or IN. 46 After IP inoculation, peak viremia occurred by day 2 PI, and peak virus titers in the liver, spleen, and especially the brain were seen at day 4 PI. Infected hamsters suffered weight loss and died between days 3 and 5 PI, depending on the route of inoculation and the dose of virus used. The VR-70 virus reportedly infected neurons in the brain but not astrocytes. Otherwise, neither the clinical signs of disease nor the pathological changes in the brains of infected hamsters were described in this report.
Pathology of the Nonhuman Primate Model of WEE
Pathology studies of NHPs with WEE viruses are limited. 65,97 In a recent study using cynomolgus macaques, 65 the ID50 of aerosolized CBA-87 strain of WEEV was determined to be 6.25 log10 pfu. Affected macaques developed fever between days 4 and 5 PI and later showed reduced appetite, decreased behavior, and tremors. Virus could not be detected in the blood or throat swabs of these macaques throughout the course of infection or in tissues other than the CNS from the animals that succumbed to infection. The main clinicopathological findings were leukocytosis, due mainly to neutrophilia, and hyperglycemia in some monkeys. Necropsies were performed on 3 of the monkeys in this study. Gross findings were limited to meningeal congestion, and the histological lesions were characterized as nonsuppurative meningoencephalomyelitis. The key microscopic changes reported in these animals were expansion of the Virchow-Robin’s spaces of the brain and some spinal cord sections by lymphocytes and monocytes; foci of necrosis containing lymphocytes, microglia, and some neutrophils; discrete glial nodules; and areas of demyelination. WEEV antigen was limited to the brain and spinal cord but was reportedly abundant within foci of inflammation and demyelination. Specifically, WEEV antigen was evident in microglial cells and neurons, including Purkinje neurons and motor neurons in the spinal cord. The authors of this study theorized that the presence of virus in the CNS, combined with their inability to find WEEV in the blood or extraneural tissues of these aerosol-infected monkeys, was evidence of neuroinvasion via the olfactory nerves.
Summary and Conclusions
The pathogenesis of alphavirus encephalitis, even when considered as a single entity, is complicated. Based on limited data from human cases and information from numerous animal studies considered to replicate presumed features of the human disease, the course of alphavirus encephalitis is characterized by 3 fundamental phases or processes. These are an initial peripheral phase of virus replication and spread, the process of neuroinvasion per se, and a terminal CNS phase in which virus spreads within the brain in particular, infects neurons primarily, and results in a neurodegenerative state that is often fatal. Complicating any discussion of animal models of alphavirus encephalitis, as we have noted throughout this review, is the point that the susceptibility of any particular animal species to any of the encephalitic alphaviruses depends on the specific strain of virus involved; the species, strain, and age of animal infected; and the dose and route of virus used for inoculation. Because significant gaps in our knowledge of the alphaviruses remain, regarding both human infections and animal models, it is also challenging to reach solid conclusions based on these studies and therefore gauge what animal models might be appropriately applied to the US Food and Drug Administration’s Animal Rule. Nonetheless, a few conclusions from these collective studies relevant to the pathogenesis of alphavirus encephalitis and suitable animal models seem reasonable to discuss.
Mice generally are susceptible to infection by virulent strains of VEEV, EEEV, and WEEV administered by a number of routes, and these animals typically develop lethal encephalitis in which massive infection of neurons is a critical feature. In contrast, NHPs less frequently develop clinical illness, manifest neurological infection, and ultimately succumb to disease with all 3 viruses. Like the mouse model, CNS infections in NHPs due to all 3 viruses involve neuronal infection, although not always to the same degree as in mice. Other animal models of VEE, EEE, and WEE may fall anywhere along a spectrum from no apparent susceptibility to lethal infection in which neuronal tropism and encephalitis are the primary consequences of CNS disease. Because neuronal damage appears to be an important contributing factor in human cases of VEE, EEE, and WEE, a variety of animal models might apply to the studies that are still needed to better understand the nature of viral targeting of neurons and the specific mechanisms that lead to neuronal damage.
The process of neuroinvasion itself is key to understanding the pathogenesis of the alphavirus encephalitides. Mice in particular show consistent neuroinvasion by alphaviruses, especially virulent strains of VEEV. At least for VEEV in mice, neuroinvasion following viremia has been shown to occur primarily in the olfactory region of the brain via the fila olfactoria. Whether this is true in other species with VEE, including NHPs and humans, is not certain. The pathway of neuroinvasion by EEE and WEE viruses is even less clear. It is clear, however, that neuroinvasion by VEEV in human infections occurs much less effectively than in the mouse. The NHP model, which also demonstrates limited neuroinvasion by VEEV, therefore better approximates the human condition and presumably shares host factors that limit the passage of virus into the brain. So it seems that NHP models should be used in future studies to better understand the process of neuroinvasion by alphaviruses. Such a strategy, however, must be weighed against the cost, availability, space, and ethical issues related to the use of NHPs in research. An alternate approach might involve the careful application of particular combinations of virus strain and animal species and age to recapitulate a degree of neuroinvasion like that evident in humans. For example, mice inoculated with attenuated strains of VEEV show a degree of neuroinvasiveness more like the human condition due to virulent VEEV and may therefore be used to investigate at least some aspects of the process of neuroinvasion. In other words, virulent viruses appear to represent viral factors that contribute to neuroinvasion, whereas attenuated strains may highlight host factors that prevent this process. Understanding the virus and host factors that mediate neuroinvasion is necessary to develop needed medical countermeasures, including safe and effective vaccines and perhaps even nonimmunological measures that might inhibit neuroinvasion.
The process of neuroinvasion by aerosolized alphaviruses in particular, a key consideration in terms of biowarfare and bioterrorism, involves a more direct conduit into the brain via the olfactory tract. Relevant to developing countermeasures, mouse studies have been important in demonstrating that IgA levels are significant in terms of limiting or preventing neuroinvasion by aerosolized VEEV. Mice will likely be relied on as an important model for at least early efficacy studies of future alphavirus vaccines intended to prevent infection by aerosolized viruses. But NHPs will also likely be required for these kinds of vaccine studies given their anatomic and immunological similarities to humans.
Infection of neurons in different regions of the brain is a key feature of alphavirus encephalitis in humans and in animals. Specific mechanisms of neuronal damage by VEEV, EEEV, and WEEV have not been as well studied as they have for the related alphavirus Sindbis. Nor have the mechanisms of interneuronal transmission of the alphaviruses been investigated in detail. These aspects seem fruitful areas for further research. In addition to viral factors of neurovirulence, a variety of host factors likely contribute to neurodegeneration in fatal cases of alphavirus encephalitis. These include immune and inflammatory features, factors related to astrocyte function, vascular factors, and even proinflammatory molecules produced by neurons themselves. A thorough understanding of the mechanisms by which neurodegeneration occurs could provide avenues to develop therapeutic countermeasures. Some of these mechanisms have already been studied in some detail in the mouse model of VEE, but similar studies in NHPs are notably lacking. This is unfortunate, because macaques infected with VEEV typically show less extensive neuronal infection and damage than either mice infected with VEEV or NHPs infected with EEEV or WEEV. Therefore, the macaque model of VEE appears to represent a condition in which host factors may contribute to severe neurological disease, presumably similar to humans with VEE. Much research is needed to characterize both human and animal infections by VEEV, EEEV, and WEEV to determine appropriate animal models of alphavirus encephalitis that can be used under the US Food and Drug Administration’s “Animal Rule” for the testing of medical countermeasures to VEE, EEE, and WEE.
Footnotes
Acknowledgements
The views, opinions, and/or findings contained herein are those of the authors and should not be construed as an official Department of Army position, policy, or decision unless so designated by other documentation. All USAMRIID studies described in this review involved protocols approved by the Institute Animal Care and Use Committee, and research was conducted in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals at the times the studies were conducted. Experiments involving animals adhered to principles stated in the Guide for the Care and Use of Laboratory Animals, National Research Council. USAMRIID is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. Previously unpublished pathology information contained in this review were generated by us from archived specimens of USAMRIID studies designed and conducted by Dr. Mike Parker, Dr. Jo Lynne Raymond, Dr. Doug Reed and Dr. Max Teehee. This review received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
The authors declared that they had no conflicts of interest with respect to their authorship or the publication of this article.
The authors declared that they received no financial support for their research and/or authorship of this article.