
Abstract
The ubiquitously expressed chloride channel 7 (CLCN7) is present within the ruffled border of osteoclasts. Mutations in the CLCN7 gene in humans (homologous to murine Clcn7) are responsible for several types of osteopetrosis in humans, and deficiencies in CLCN7 can present with retinal degeneration and a neuronal storage disease. A previously reported Clcn7 –/– mouse showed diffuse osteopetrosis accompanied by severe retinal and neuronal degeneration. In contrast, the authors produced a novel Clcn7 –/– mutant where mice did not develop osteopetrosis but still developed lethal neural and retinal degeneration. In these mice, there was a rapid progressive loss of the outer nuclear layer and photoreceptor layers of the retina. Laminar degeneration and necrosis of neurons in layers IV and V of the cerebral cortex and in the CA2/CA3 regions of the hippocampus were associated with intraneuronal accumulations of autofluorescent granules (periodic acid–Schiff positive). The extensive reactive gliosis was always associated with the accumulation of intraneuronal cytoplasmic material. The authors found, through quantitative real time polymerase chain reaction analyses, that an alternate Clcn7 transcript (previously identified only in bone marrow) showed minimal expression in the brain and eye but moderate expression in bone, which correlates with rescue of the osteopetrotic phenotype in the face of continued retinal and neuronal degeneration. Findings in this knockout mouse model prove that osteopetrotic compression of the brain is not responsible for neuronal and retinal degeneration in CLCN7-deficient mice; rather, they suggest that neurotoxicity is most likely due to lysosomal dysfunction as a result of the functional lack of this chloride channel in the central nervous system and eye.
Osteopetrosis is characterized by increased bone mass owing to deficient osteoclastic bone resorption. 45 For effective resorption of bone, differentiated osteoclasts must form a sealing zone that isolates resorption lacunae from the extracellular environment, and they must develop a ruffled membrane that secretes acid and proteases. 47 Animal models of osteopetrosis have contributed much to our understanding of the functions and roles of osteoclasts in normal bone biology. 47 Mutant rodents with osteopetrosis enabled the discovery of the hematological origin of osteoclasts and the potential utility of bone marrow transplantation in treating osteopetrosis. 50,51 Knockout mice have proven invaluable in clarifying many of the processes involved in the differentiation and function of osteoclasts (reviewed in Del Fattore 9 ).
Osteopetrosis most often results from deficiencies in proteins involved in the acidification of resorption lacunae. In this process, the first critical protein is carbonic anhydrase type II, an enzyme that catalyzes the hydration of carbon dioxide to carbonic acid. A second enzyme, the vacuolar H+-ATPase (V-ATPase), pumps protons from carbonic acid across the cell membrane. Mutations in the a3 subunit of the V-ATPase enzyme are responsible for approximately 50% of human autosomal recessive osteopetrosis, 39 and the oc/oc osteopetrotic mouse has a large deletion in the homologous V-ATPase gene. 35 The third protein, chloride channel 7 (CLCN7), functions as a Cl–/H+ exchanger across the same membranes. 25 The ubiquitously expressed CLCN7 is located in late endosomes and lysosomes 21 and in the ruffled membrane of osteoclasts. 25 The efficiency of the V-ATPase proton pump in acidifying extracellular and intracellular compartments, including endosomes and secretory vesicles, depends on CLCN7 to maintain electroneutrality by transferring negatively charged chloride ions to balance the positively charged protons. 1 Because osteoclasts that lack functional CLCN7 fail to acidify resorption lacunae and cannot degrade bone, mutations in the human chloride channel gene CLCN7 result in several forms of osteopetrosis, ranging in severity from asymptomatic or mild symptoms in patients with autosomal dominant osteopetrosis 49 to a severe autosomal recessive form: malignant infantile osteopetrosis (autosomal recessive osteopetrosis). CLCN7 mutations are also associated with the relatively mild forms of intermediate autosomal recessive osteopetrosis. 5
Neurological defects and blindness can develop in humans and mice with mutations in CLCN7. 12,22,25 Osteoclastic bone remodeling is necessary for preventing stenosis of bone foramina that would otherwise compress optic, vestibulocochlear, and other cranial nerves. 9 Some deafness and blindness associated with osteopetrosis have been attributed to nerve compression caused by overgrowth of bone. 4,44 For example, the visual loss in patients with autosomal dominant osteopetrosis appears to be secondary to the bone phenotype. 49 In other cases, the visual loss was associated with a primary retinal and neuronal degeneration, 33 and it has been linked to mutations in CLCN7 or the gene for an associated protein, OSTM1. 12,22,25,26
To elucidate the cellular and physiological roles of CLCN7, we generated a novel Clcn7 –/– mutant mouse line using a different deletion strategy than that used previously. Other groups previously reported that inactivation of the Clcn7 gene in mice eliminated the acid-secreting ability of osteoclasts and resulted in mice that had a gray coat; were stunted and short-lived; and developed chondrodysplasia, osteopetrosis, failure of tooth eruption, and retinal and neuronal degeneration. 25,26 In contrast, our Clcn7 –/– mice exhibited normal osteoclastic function (as shown by the absence of osteopetrosis) and normal coat color. Nevertheless, our knockout mice were smaller than wild-type littermates and still died at approximately 6 weeks with severe retinal and neuronal degeneration. We report here evidence indicating that the absence of osteopetrosis in our knockout mice was due to the bone-specific transcription of a Clcn7 splice variant. Our findings show that osteopetrotic compression of the brain and optic nerve is not involved in neuronal and retinal degeneration and that CLCN7 deficiency is toxic to specific cell populations in the retina and brain.
Materials and Methods
Generation of Mutant Clcn7–/– Mice
The Clcn7 conditional targeting vector was derived using the Lambda KOS system. 52 The vector was designed to remove 455 base pairs of genomic DNA overlapping the ATG of the first coding exon from position 25270362 to position 25270816 of chromosome 17, based on the July 2007 genome assembly at University of California, Santa Cruz, and NM_011930.3. Conditional mice were subsequently crossed with mice containing a Protamine-Cre transgene 30 resulting in germline excision of the region, which was confirmed by Southern blot hybridization. Mice were genotyped by polymerase chain reaction (PCR). The wild-type allele yielded a PCR product of 583 base pairs, and the mutant allele, a PCR product of 230 base pairs, with primers F: GCACCGATGATAGATCACGT and R: GCCTTGTATGTGTAAACCCTG. All analyzed mice were maintained on a mixed genetic background (129S5/SvEvBrd and C57BL/6J) at the animal facility of Lexicon Pharmaceuticals, Inc (Animal Lovers Against Animal Cruelty accredited). Studies of gene-disrupted or humanized animals were performed with wild-type littermates as controls. The homozygous F2 knockout mice used in phenotyping studies were produced by intercrossing the F1 heterozygous knockout (–/+) offspring of chimeric founder parents. All mice were housed in a barrier facility at 24°C on a fixed 12-hour light and 12-hour dark cycle and were fed rodent chow ad libitum (No. 5001, Purina, St Louis, MO). Procedures involving animals were conducted in conformity with the Institutional Animal Care and Use Committee guidelines, which are in compliance with state and federal laws, and with the standards outlined in the National Research Council’s Guide for the Care and Use of Laboratory Animals.
Histology and Immunohistology
Immediately after euthanasia, Clcn7 –/– mice and age-matched control mice were fixed by cardiac perfusion with Bouin fixative, and tissues were collected and immersed in 10% neutral buffered formalin for an additional 48 hours, except for the eyes, which were removed and fixed by immersion in Davidson’s fixative (Poly Scientific, New York, NY) overnight at room temperature. All tissues were embedded in paraffin, sectioned at 4 μm, and mounted on positively charged glass slides (Superfrost Plus, Fisher Scientific, Pittsburgh, PA) and stained with hematoxylin and eosin (HE) for histopathologic examination. Selected replicate sections were stained with periodic acid–Schiff (PAS), Luxol fast blue, Sudan black B, acid-fast, toluidine blue, Alcian blue, Nissl, and Bielschowski silver stains. In addition, unstained sections were mounted with DAPI and examined by fluorescent microscopy for brain autofluorescence. Exposure-matched digital images were taken from comparable regions of wild-type and knockout brains. Selected sections of brain were immunostained for glial fibrillary acid protein (GFAP) and ubiquitin. For immunohistochemistry, 4-μm sections were first deparaffinized in xylene and then rehydrated through graded alcohols to phosphate buffered saline (PBS); furthermore, endogenous peroxidase activity was blocked by incubation in 3.0% hydrogen peroxide in PBS for 5 minutes. Epitope retrieval was performed by immersing slides for 20 minutes in a preheated (95–100C°) citrate buffer (pH 6.0) solution. Potential nonspecific binding sites were blocked with 20% normal goat serum in PBS. Primary antibodies for immunochemistry were rabbit anti-GFAP (Dako, Carpinteria, CA) and rabbit anti-ubiquitin (Dako), which were diluted at 1:1,000 and 1:400, respectively, in PBS and applied for 1 hour at room temperature. After rinsing, sections were incubated for 1 hour in the biotinylated goat anti-rabbit immunoglobulin G secondary antibody (Dako) diluted 1:500 in PBS. Bound antibodies were detected by a horseradish peroxidase–streptavidin method, with 3,3′-diaminobenzidine as chromogenic substrate based on the manufacturer’s instructions (Dako).
MicroCT
Cortical thickness of the femoral midshaft (20-μm voxel size) and cancellous bone volume (16-μm voxel size) in the distal femoral metaphysis were determined with a Scanco μCT40 scanner (Scanco Medical AG, Basserdorf, Switzerland). A threshold of 240 was used for evaluation of all scans. Twenty slices (0.4 mm) were analyzed in the midshaft femur. Dual energy x-ray absorptiometry scans were performed with a PIXImus2 Mouse Densitometer (version 2.10, GE Medical Systems, Madison, WI).
Real-time quantitative reverse transcription PCR was conducted for expression of Clcn7 variants in wild-type and homozygous mouse tissues: RNA was extracted from brain, muscle, liver, lung, heart, kidney, and bone (both with marrow intact and with marrow flushed out with saline) from 2 wild-type mice and 3 knockout mice. Total RNA was extracted with the Invitrogen Trizol RNA purification kit (catalog No. 155596-018). An OD 260/280 measure was taken of 1:20 dilutions of the resuspended RNA to determine yield and purity. For the reverse transcription reactions, 50 μl of RNA was used for the plus reverse transcription, and 25 μl for the minus reverse transcription, using the high-capacity cDNA archive kit (ABI 4322171). A 2-μl aliquot of the cDNA was subsequently used in a 20-μl reaction mix and run in a 384-well format, with default cycling conditions for 50 cycles with duplicate sample reactions. The primers used to detect the common known variant with exon 1 were F1: 5′ CGG GTC ACG GGA ATG CT and R1: 5′ TGG CCC ACT GAG GAC CAA with the TaqMan probe P1: 5′TGC CGG CTG TTC TTG TTG AG. To detect the variant transcript, annotated as being from the bone macrophage, starting with alternate exon A (BY743288), primers were designed within exon A: FA: 5′ GAG AGG TGG CAG TGT GTG TTT G and RA: 5′ GAG GTG TCT CTG GTG CGA TTC with TaqMan probe PA: 5′AGT CTT TAA ATG CAG GGC ATT CTT ACA GGC C. The expression level of both and 18S in the different tissues was quantified on the basis of the standard curve method via real-time quantitative reverse transcription PCR on an ABI PRISM 7900 sequence detector using the ABI 18S rRNA kit (ABI 4319413E). The relative expression of mRNA in the different tissues was normalized to the amount of 18S rRNA in the same sample (Fig. 1 ).
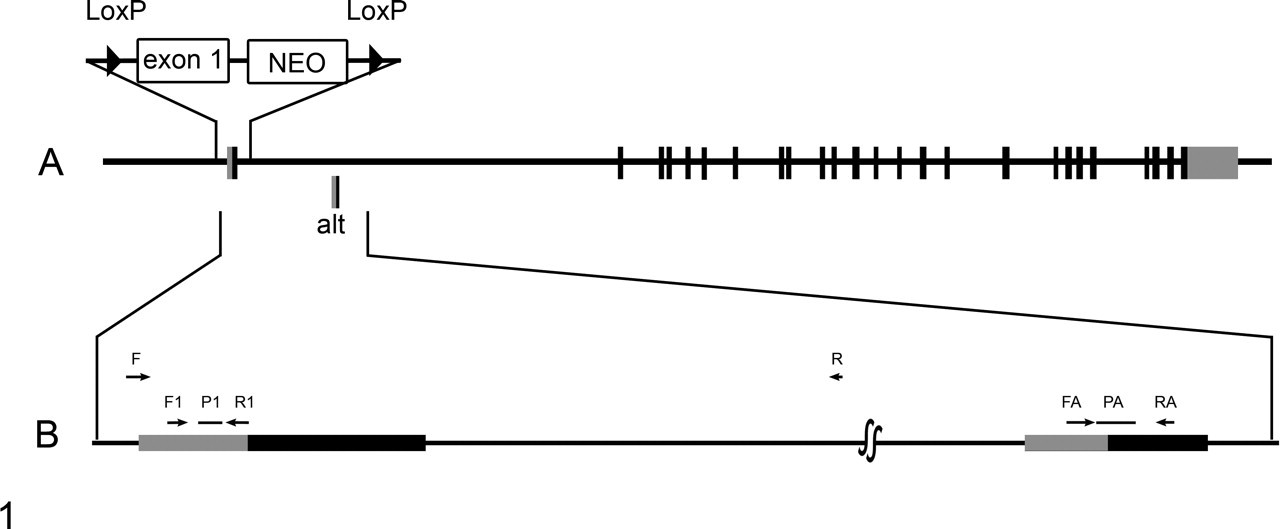
A, the gene-targeting strategy in which exon 1 was replaced with a targeting vector containing a NEO selection cassette flanked by LoxP sites. The relative position of the alternative start site is shown (alt). B, an expanded segment of the gene to illustrate the relative locations of the quantitative real time polymerase chain reaction primers used to amplify the standard exon 1 transcript (F1 and R1 primers, P1 probe) and the alternate exon 1 transcript (FA and RA primers, PA probe). Boxes show the 5′ untranslated (gray) and translated (black) segments.
Results
Wild-type, heterozygous, and homozygous F2 littermates were produced in a normal 1:2:1 Mendelian ratio; of 463 offspring, there were 113 wild-type, 241 heterozygote, and 109 homozygote mice (χ2 = 0.651). No differences in hair color were noted in agouti or black mice, but Clcn7 –/– mice were easily distinguishable from heterozygous and wild-type littermates at 4 weeks of age by their stunted growth (data not shown). Neurological signs first became evident from 3 weeks of age, initially appearing as mild tremors and stiffness and progressing in severity to tonic seizures and respiratory arrest in terminal stages. All Clcn7 –/– mice either died or were euthanized before reaching 7 weeks of age.
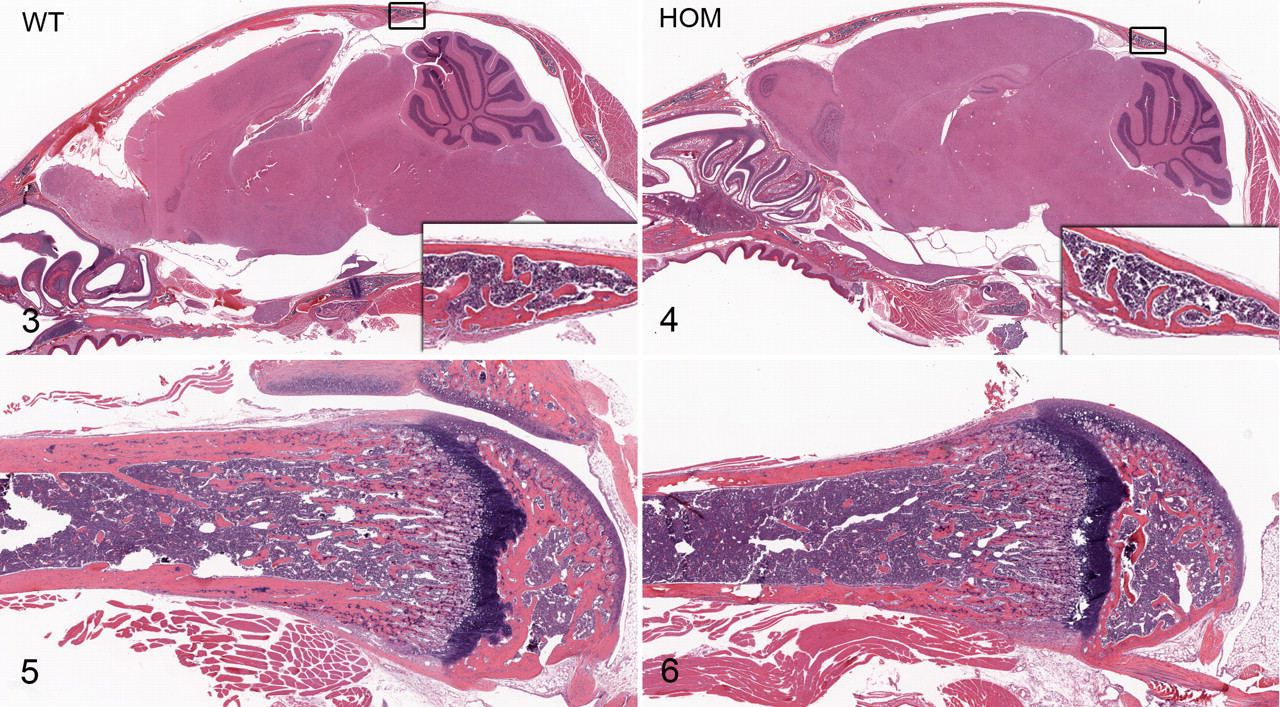
No skeletal or bone malformations were detected in our Clcn7 –/– mice. Both dual energy X-ray absorptiometry and micro–computed tomography analysis of bones showed similar cortical and trabecular bone volume measurements in the Clcn7 –/–mice compared with wild-type littermates (Fig. 2 ). In addition, the bones of the skull and limbs showed normal morphology on histologic examination (Figs. 3–6), and osteoclasts did not appear to differ in number or morphologic appearance from those in wild-type littermates.
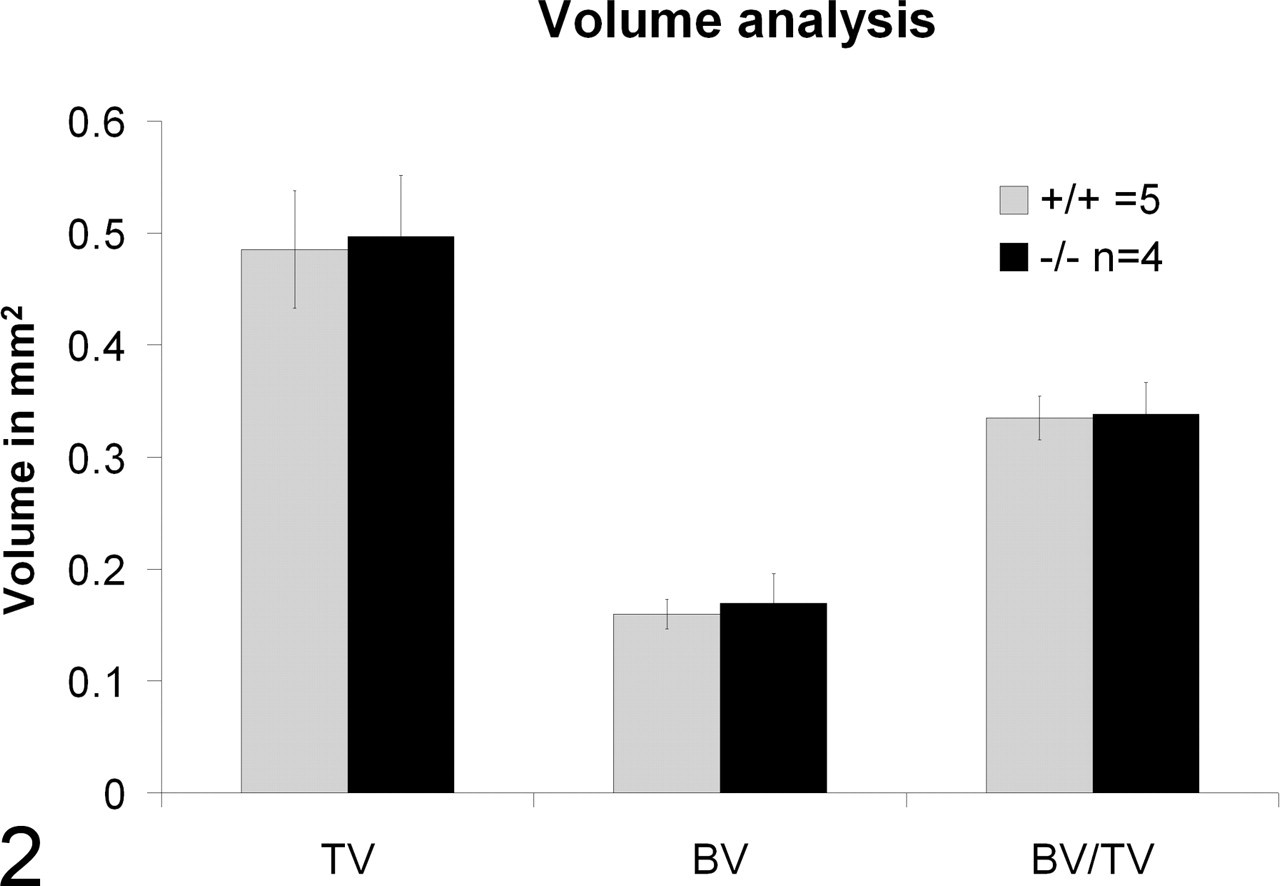
Femoral bone micro computed tomography findings: Histogram shows similar trabecular volume (TV), cortical bone volume (BV), and cortical/trabecular bone ratios in wild-type (+/+) and knockout (–/–) mice.
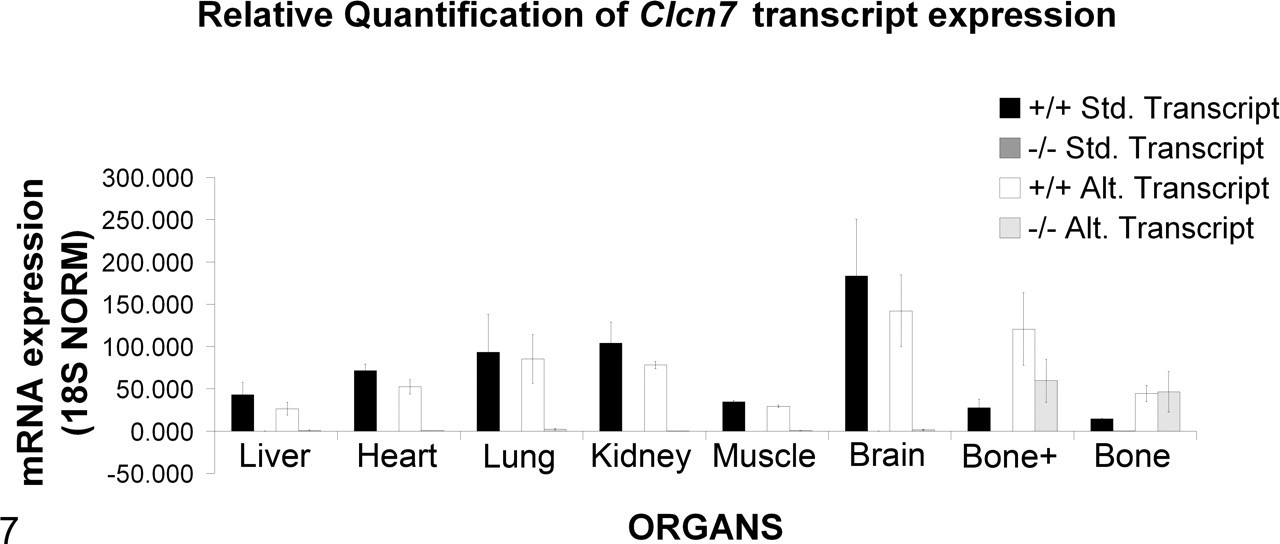
Quantitative reverse transcription polymerase chain reaction for expression of standard and variant Clcn7 transcripts in wild-type and homozygous mouse tissues. Expression of the standard transcript was detected in all wild-type tissues analyzed. In contrast, deletion of exon 1 resulted in absence of the standard transcript originating at exon 1 in all tissues analyzed in our Clcn7 –/– mice. Similarly, primers targeting the alternate exon A showed that the alternate transcript was present in all the tissues analyzed in the wild-type mice, including the bone. However, in Clc7 –/– mice, the alternate transcript was detected in only the bone (with and without marrow). Bone+, bone with marrow.
The absence of osteopetrosis in our mouse line was surprising, but because our gene-targeting strategy was different from that used in previous reports, we asked if alternate transcripts could be the underlying reason for the difference in phenotype. A search of available databases revealed the existence of an alternative transcript predicted by Gnomon (National Center for Biotechnology Information’s eukaryotic gene prediction program), based on an expressed sequence tag obtained from mouse bone macrophage cells (GenBank: BY743288.1). This transcript uses an alternate start exon designated as exon A. We performed quantitative real time polymerase chain reaction PCR analysis of several tissues, including liver, heart, lung, kidney, muscle, brain, bone with marrow (bone+), and bone without marrow (bone). Our results show that deletion of exon 1 resulted in absence of the transcript originating at exon 1 from all the tissues analyzed in our Clcn7 –/– mice, although it was present in all wild-type tissues analyzed (Fig. 7 ). Primers targeting the alternate exon A showed that this transcript was present in all the tissues analyzed in the wild type, including the bone (Fig. 7). However, in our Clcn7 –/– mice, the alternate transcript was detected in only the bone (with or without marrow). It seems likely that this alternative transcript is responsible for rescuing the bone phenotype in these mice. The alternate transcript was not present in any other tissues examined from Clcn7 –/– mice. Such tissue-specific regulation of variant expression is not unusual in mammals, and further mutation analysis will be required to fully understand the details of tissue-specific regulation of CIC7.
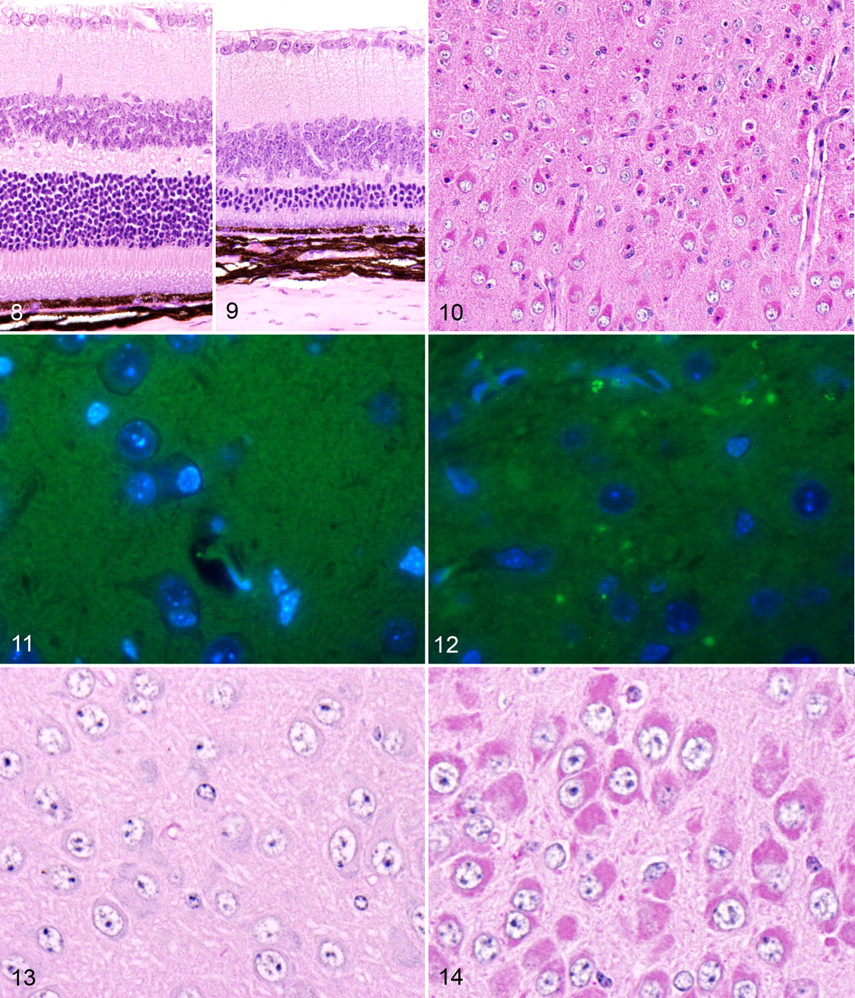
In contrast to the normal findings in bone, severe lesions were present in the brain and eyes of our Clcn7 –/– mice. Selective regional neurodegeneration of specific subpopulations of neurons and retinal cells was present in the central nervous system and eye, respectively. In contrast to normal retina (Fig. 8 ), homozygous mice showed severe thinning of the outer nuclear layer and complete loss of inner and outer segments of the photoreceptor layer (Fig. 9 ). There was only minimal thinning of the inner nuclear layer (bipolar cell layer), and cellularity within the ganglion cell layer appeared essentially normal. A few macrophages were present in the subretinal space and the retinal pigmented epithelium was intact, although some retinal pigmented epithelium cells were mildly hypertrophic. Pigmentation of the retinal pigmented epithelium, iris, and choroid, as well as hair, was normal in black and agouti mice.
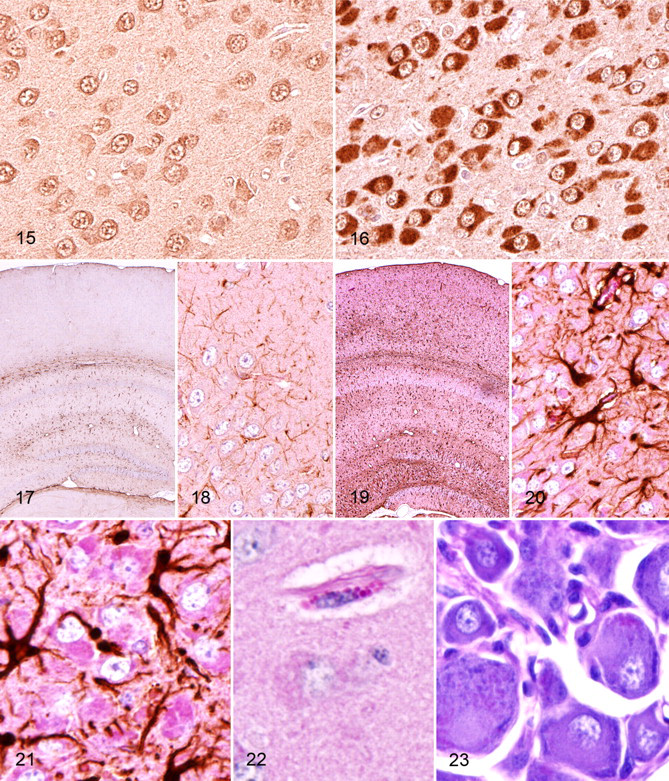
At 3 weeks of age, the hallmark lesion was laminar neuronal necrosis in the cerebral cortex (Fig. 10) with focal areas of neuronal degeneration in the hippocampus. By 6 weeks of age, the laminar pattern of necrosis in the neocortex was obscured by progression of the disease, and in these terminally ill mice, there was widespread neuronal necrosis accompanied by neuronophagia, microglial activation, and astrogliosis in the neocortex. Terminally, cortical layers IV and V were almost entirely absent, and the other cell layers of the neocortex appeared to be reduced in thickness in knockout mice compared with wild-type littermates. The CA2 and CA3 regions of the hippocampus had undergone massive neuronal death by 6 weeks of age. Degeneration and necrosis of hippocampal pyramidal neurons was initially focused in the CA3 region and so progressed over time to include the CA2 region. In contrast, neurons in the dentate gyrus and CA1 region of hippocampus were largely spared and showed only relatively mild neuronal degeneration and accumulations of granular material. Standard HE staining of brains of knockout mice demonstrated a striking accumulation of eosinophilic cytoplasmic granules in most cortical and hippocampal neurons. Almost all intact neurons in all layers of the cerebral cortex and in the CA2 and CA3 areas of the hippocampus were distended by myriad closely packed, small (1–2 μm) deeply eosinophilic intracytoplasmic granules, which often displaced the nucleus and/or Nissl substance to the periphery. Examination of these same areas with fluorescence microscopy revealed broad-spectrum autofluorescence of some of the larger granules within neurons and the neuropil in knockout mice only (Figs. 11, 12). Special staining procedures showed that the cytoplasmic granules stained positively with PAS (Figs. 13, 14), and immunostaining showed that the storage material stained strongly for ubiquitin (Figs. 15, 16), suggesting that the granules represent autophagosomes that contain denatured but incompletely digested ubiquitinated proteins following fusion with functional lysosomes. In HE-stained sections, rare degenerating neurons and swollen axonal processes were in other areas of the brain, but visible pathological changes were essentially absent in the thalamus, hypothalamus, brainstem, and spinal cord. However, small amounts of PAS-positive granular storage material were detected in virtually all areas of the midbrain, cerebellum, and spinal cord and even in peripheral ganglia. In all locations, storage material failed to stain positively with Luxol fast blue, Sudan black B, acid-fast, toluidine blue, Alcian blue, Nissl, and Bielschowski silver stains.
Detectable microgliosis and satellitosis were generally restricted in HE-stained sections to cortical and hippocampal areas with severe neurodegeneration, but increased reactive gliosis was demonstrated diffusely in the central nervous system by immunohistochemical staining with anti-GFAP antibody. In wild-type brains, intense GFAP staining was restricted in distribution and most often concentrated near white tracts, and the fine cytoplasmic processes of normal glial cells was highlighted by GFAP staining (Figs. 17, 18). In contrast, GFAP immunostaining in knockout mice was more intense and widely dispersed (Fig. 19 ), and it appeared to precede obvious neurodegeneration in most areas of the central nervous system. GFAP immunostaining highlighted the increased cytoplasmic volume and thickened cytoplasmic extensions of reactive glial cells throughout the central nervous system (Fig. 20 ). Nevertheless, GFAP staining was clearly most diffuse and intense in the severely affected areas of the cortical gray matter and hippocampus, suggesting a strong correlation between the extent and intensity of GFAP-positive staining and the amount of storage material in neurons and consequent neuronal degeneration. Even in areas where neuronal loss was not obvious, the accumulation of intracytoplasmic storage material was accompanied by prominent hypertrophic GFAP+ astrocytes (Fig. 21 ), and this astroglial hypertrophy (representing activation) appeared to be more widespread in brains of 6-week-old mice than in 3-week-old mice (data not shown). In severely affected areas, PAS staining highlighted the large cytoplasmic granules that accumulated in scattered perivascular and meningeal macrophages (Fig. 22 ). Even though no degenerative lesions were apparent in the brainstem, spinal cord, and peripheral ganglia of symptomatic knockout, small amounts of PAS-positive storage material were present in some motor neurons and peripheral ganglionic neurons (Fig. 23 ). However, peripheral nerves appeared to be unaffected, and there was no evidence of motor neuron loss or neurogenic muscular atrophy. Although storage material accumulated throughout the central nervous system, none was detected in other extraneuronal tissues and visceral organs.
Discussion
Mutations in the intracellular chloride channel gene CLCN7 are implicated in several types of human osteopetrosis, including some cases with malignant infantile osteopetrosis accompanied by retinal and neuronal degeneration, leading to blindness and early death. 22,25,26 For many years, the neurodegeneration observed in various types of osteopetrosis was attributed to the progressive compression of brain, cranial nerves, spinal cord, and blood vessels by osteopetrotic bone. Indeed, in some forms of osteopetrosis, the constriction of the foramina through which optic and vestibulocochlear nerves pass does contribute to retinal atrophy and deafness, respectively. 40 However, approximately 10% of cases with malignant infantile osteopetrosis develop a rapidly progressive retinal and neuronal degeneration that is apparently not secondary to osteopetrotic bone compression. Instead, these cases develop a neurodegenerative phenotype that has important similarities to the lysosomal storage disease known as neuronal ceroid lipofuscinosis (NCL). 40
The absence of bone lesions in our Clcn7 –/– mice was unexpected given the severe osteopetrosis previously associated with inactivating mutations of this gene in humans and in another line of Clcn7 –/– mice. 25 Despite the absence of osteopetrosis, our Clcn7 –/– mice developed the same lesions of severe progressive neurodegeneration and retinal degeneration previously reported in osteopetrotic Clcn7 –/– mice. 25 Interestingly, the phenotype of our knockout mice was similar to that reported in Clcn7 –/– mice that expressed transgenic CLCN7 in the bone under the control of tartrate-resistant acid phosphatase; although these transgenic knockout mice had normal osteoclastic function and normal bone structure, their average life span was extended by only 3 weeks, and they died with progressive neuronal and retinal degeneration. 22 Upon closer examination, we concluded that the strikingly different bone phenotype in our Clcn7 –/– mice compared to those described previously stemmed from the two different Clcn7 –/– gene deletion strategies used in producing the knockout mice.
In our line, exon 1 was replaced by the selection cassette, whereas Kornak et al deleted exons 3–7. 25 We discovered that our knockout strategy allowed the expression of an alternate transcript using the alternate exon A. Possible explanations for the differential expression of these variants in mice and humans are based on the genomic analysis of human and mouse splice variants. First, we noted that the mouse expressed sequence tag database identified a unique alternate exon A in bone macrophage cells (BY743288). The splice variant transcribed with the alternate exon A replaces the first 45 amino acids of the primary transcript with a novel 25 AA sequence, but it is in all other respects identical to the ubiquitously expressed form. In our Clcn7 –/– mice, deletion of exon 1 would leave the bone macrophage splice variant intact, whereas in the previously reported knockout, both variants are deleted, probably accounting for the phenotypic differences. The characterization and quantification of this variant transcript demonstrated that it had a distinct expression profile as compared with the normal full-length transcript. The alternate transcript appeared to be more highly expressed in wild-type bone compared with the full-length transcript, whereas its expression in most wild-type tissues was somewhat less than the full-length transcript. In our Clcn7 –/– mice, the alternate transcript represented the only significant expression of Clcn7 and was found in bone only; the full-length transcript was completely abrogated by deletion of exon 1. Thus, the phenotype displayed by our knockout mice could reasonably be attributed to expression of this alternate transcript in bone. Given the normal hair color of our Clcn7 –/– mice, this alternate transcript would likely be expressed in skin, but gene expression in skin was not evaluated in this study.
Upon review of the human genome databases, a human ortholog of the mouse alternate exon A may not exist. This finding suggests that orthologous mutations in CLCN7 that might prevent osteopetrosis without averting neurodegeneration may not occur in humans. Alternatively, multiple known human splice variants (AceView) could encode other isoforms (http://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/av.cgi?c=geneid&org=9606&1=1186).
Regardless, in our mice, expression of CLCN7 was likely retained in bones (osteoclasts), resulting in a normal bone phenotype. In addition, our findings prove that CLCN7-related neuronal and retinal degeneration occurs independently of any physical compressive effects of osteopetrotic bone. The exquisite susceptibility of photoreceptors and neurons to the deleterious effects of CLCN7 deficiency suggest strongly that CLCN7 blockers intended to therapeutically reduce bone resorption 34 need to be carefully tested for deleterious side effects in the eye and brain.
The lesions in the central nervous system have many characteristics of a lysosomal storage disorder. Many heritable enzyme deficiencies are accompanied by the accumulation of undegraded substrates within lysosomes, and lysosomal storage defects frequently show neurological involvement. In our mice and as reported previously, 22 neurodegeneration in Clcn7 knockout mice is associated with the accumulation of autofluorescent material in neurons. The lysosomal localization of CLCN7 suggests an important role for this chloride channel in lysosomal functions and supports the classification of this neurodegenerative disease as a lysosomal storage disorder. In addition, the pathological changes in Clcn7 knockout mice have important similarities to those occurring in the NCLs, which are lysosomal storage diseases characterized by the intracellular accumulation of autofluorescent storage material in the brain and other tissues.
NCLs include a variety of recessively inherited neurodegenerative diseases characterized by lysosomal storage of autofluorescent, PAS-positive, and Sudan black B–positive granules in neurons and other cell types. 14 Interestingly, the NCLs were not recognized as lysosomal storage disorders until relatively recently, when NCL genes were found to encode proteins that localize to the endosomal/lysosomal compartment. 28 Mutations in at least 7 genes underlie the multiple human and animal forms of NCL, 15 and animal models have proven invaluable in elucidating the pathogenesis of NCLs (see review 19 ). Mouse models with deficiencies in the same genes involved in human NCL (cathepsin D, CLN1, CLN2, CLN3, CLN5, CLN6, CLN8 [CLN: for ceroid lipofuscinosis, neuronal]) develop progressive neurodegenerative phenotypes that have been useful in discovering the molecular mechanisms underlying NCL gene disorders; 8 furthermore, knockout mouse models showed the selective nature of neurodegeneration, provided evidence that glial responses precede neuronal loss, and identified the thalamus as an early pathological target in NCL disease. 8
More recently, other NCL-like diseases have been identified, which also involve lysosomal or lysosome-related gene products. Mouse models of NCL-like disease (reviewed in Cooper et al 8 ) include knockouts of Clcn7, 22 Clcn3, 53 PPT1, and PPT2 (for palmitoyl-protein thioesterase 1) 13 and the cathepsin D, 23 F, 43 and B/L proteases. 24 The lack of lysosomal cathepsins induces extensive neuron death and neurodegeneration that is preceded by an accumulation of autophagosomes. 24 Cathepsins are lysosomal proteases responsible for the degradation and turnover of proteins at acidic pH. 6 Deficiency of cathepsin D, which is a lysosomal aspartic protease, results in severe progressive NCL in infants, sheep, and mice. 20,23,38,46 The combined cathepsin B/cathepsin L–deficient mouse dies at approximately 2 weeks of age with massive brain atrophy, neuron cell death, and reactive gliosis. 11 As noted previously, the two chloride channels (ClC-3 and CLCN7) that have been linked to NCL-like disease are both located on endosomal/lysosomal membranes and are both required for efficient acidification of intracellular vesicles by providing electrical neutralization of protons that are actively pumped across membranes by V-ATPases. It appears that deficiencies in ClC-3 and CLCN7 may impair lysosomal proteolysis and result in lysosomal accumulation of metabolites. 21 The accumulation of autofluorescent intracytoplasmic material in the central nervous system and retina is the hallmark pathologic lesion in these knockout mice. However, this change is not specific to NCLs: Intracellular autofluorescent material accumulates with aging 10 and can be pharmacologically induced with compounds that block protein degradation in lysosomes such as leupeptin (a thiol proteinase inhibitor) and chloroquine (a general lysosomal enzyme inhibitor). 16,18 The pigment that accumulates as a normal part of aging is termed lipofuscin, whereas ceroid is the general term for the lipopigment that accumulates as a result of a pathological condition. 36 Nevertheless, the lysosomal location of CLCN7, the diffuse intraneuronal accumulation of intracytoplasmic autofluorescent PAS-positive granules, and the neuropathological similarities to established mouse models of NCL support a role for lysosomal dysfunction in the pathogenesis of neuronal and retinal degeneration in Clcn7 –/– mice.
The accumulation of poorly degraded substrates in neurons in many lysosomal storage diseases is associated with neuronal necrosis, and we observed the largest accumulations of PAS-positive, autofluorescent material in regions with widespread neurodegeneration. It is not known if the accumulated materials are directly toxic, but the chemical diversity of lysosomal contents in the various storage diseases suggests that common pathogenetic mechanisms are involved in neurodegeneration. Interestingly, in an early report on infantile lysosomal storage disease associated with osteopetrosis, lectin histochemistry that was used to characterize the PAS-positive storage material indicated that there is storage of carbohydrates and lipids in neurons. 2 Lectin staining with Concanavalia ensiformis agglutinin, Datura stramonium agglutinin, Griffonia simplicifolia-I, Lens culinaris agglutinin, Ricinus communis agglutinin-I, succinylated wheat germ agglutinin, and wheat germ agglutinin indicated that the storage material consisted of fucosylated N-glycosidically linked oligosaccharides containing b- and a-galactosyl residues and compounds containing N-acetyl lactosamine. 2
It is clear that some neurons are more susceptible to lysosome dysfunction (reviewed in Pivtoraiko et al
31
). The outer nuclear (photoreceptor) layer of the retina appears to be exquisitely sensitive to lysosomal dysfunction. As in many other forms of NCL, retinal degeneration in Clcn7
–/– mice developed with neuronal degeneration of specific neuronal subsets in the hippocampus and cerebral cortex. In cathepsin D and Clcn3 knockout mice,
Interestingly, the specific areas of neuronal degeneration in the brain differ in the various sforms of NCL and NCL-like diseases. In Clcn7 –/– mice, there is diffuse neurodegeneration of CA2/CA3 hippocampus and cerebral cortex, with relative sparing of CA1, whereas in Clcn3 –/– mice, the CA1 region of the hippocampus is the first area to degenerate, and cerebral cortical involvement is much milder. Although the other hippocampal regions are eventually affected in Clcn3 –/– mice, with almost complete destruction of the hippocampus at 8 weeks, 41 most Clcn3 –/– mice survive over 1 year. In these diseases, dysfunctional lysosomes are believed to induce caspase-dependent and caspase-independent cell death via oxidative stress and free radical damage. 37 Apoptotic cell death is most often considered the principal route of neurodegeneration in NCL and other central nervous system diseases, but other pathways appear to be involved. Two principal cell death pathways, as defined by morphological criteria, are type I apoptotic and type II autophagic. 7 Type I apoptotic death is regulated by proapoptotic and antiapoptotic members of the Bcl-2 family, 37 and it culminates in the activation of caspases, which are responsible for the nuclear condensation, DNA fragmentation, and cell shrinkage that are the morphologic hallmarks of apoptosis. 7
In contrast, type II autophagic death is morphologically defined by the aberrant accumulation of autophagic vacuoles in degenerating cells, which may also display some morphological features of apoptosis. 7 In addition to NCL and lysosomal storage disorders, neurodegenerative diseases such as Alzheimer, Parkinson, and Huntington are associated with the accumulation of storage proteins and so display features of autophagy and/or apoptosis. 3 Recent evidence provides support for the view that neurodegenerative processes in NCL involve activation of autophagic and apoptotic cell death pathways and that complex interrelationships exist between these two pathways. 24,29
Reactive gliosis was diffusely present in the central nervous system of our Clcn7 –/– mice and was most severe in areas containing abundant neuronal storage material and degenerating/apoptotic neurons. Although reactive gliosis is a characteristic lesion in all forms of NCL, 14 it has been unclear whether glial activation precedes or is triggered by neuronal loss. A recent report showed upregulation of markers of astrocytic and microglial activation in presymptomatic Cln3 –/– mice many months before the onset of significant neuronal loss. 32 However, with PAS staining, we discovered the presence of intraneuronal storage material throughout the central nervous system, including many regions that appeared entirely normal in HE-stained sections. Our findings indicate that although reactive gliosis is often present in areas without evidence of neuronal loss, it is always associated with evidence of lysosomal storage.
In summary, we used targeted gene disruption to generate CLCN7-deficient mice. These mice developed a progressive NCL-like disease characterized by the accumulation of intracytoplasmic PAS-positive, autofluorescent granules in neurons, which were associated with neuronal necrosis in more severely affected areas. Surprisingly, our knockout mice significantly differed from other mutant CLCN7 mice in having normal bones and hair coat color. Upon closer examination, we found that these Clcn7 –/– knockout mice (1) express an alternatively spliced transcript of the Clcn7 gene that apparently maintains normal osteoclast function and rescues osteopetrosis and normal coat color, (2) exhibit a rescued bone phenotype that correlates with transcription of the alternative Clcn7 transcript in bone, and (3) die at approximately 6 weeks of age with severe retinal and neural degeneration associated with an apparent lysosomal storage disease. Our findings in this knockout mouse model prove that osteopetrotic compression of the brain is not required for neuronal and retinal degeneration; they also indicate that lysosomal dysfunction due to CLCN7 deficiency is neurotoxic to a subset of neurons in the central nervous system and eye. Further studies with this variant form may open new avenues of research into the pathogenesis of fatal autosomal recessive storage disorders and the mechanisms of neuronal cell death.
Footnotes
Acknowledgements
We thank Kathy Henze and James Syrewicz for providing histology sections and immunohistochemistry. We also thank Dr. Tom Lanthorn for helpful discussions and support for this work.
The authors declared that they had no conflicts of interest with respect to their authorship or the publication of this article.
The authors were all employees of Lexicon Pharmaceuticals Inc., which provided financial support for these studies.