
Abstract
Cyclooxygenase (COX) catalyzes the conversion of arachidonic acid into prostaglandin H2 (PGH2), which is subsequently converted to the prostanoids PGE2, PGI2, PGF2α, and thromboxane A2. COX has 2 distinct membrane-anchored isoenzymes: COX-1 and COX-2. COX-1 is constitutively expressed in most normal tissues; COX-2 is highly induced by proinflammatory mediators in the setting of inflammation, injury, and pain. Inhibitors of COX activity include conventional nonselective nonsteroidal anti-inflammatory drugs and selective nonsteroidal anti-inflammatory drugs, such as COX-2 inhibitors. The adverse effects of COX inhibitors on the cardiovascular system have been addressed in the last few years. In general, COX inhibitors have many effects, but those most important to the cardiovascular system can be direct (through the effects of prostanoids) and indirect (through alterations in fluid dynamics). Despite reports of detrimental human cardiovascular events associated with COX inhibitors, short, long, and lifetime preclinical toxicology studies in rodents and nonrodents have failed to identify these risks. This article focuses on the expression and function of COX enzymes in normal and pathologic conditions of the cardiovascular system and discusses the cardiovascular pathophysiologic complications associated with COX inhibition.
Keywords
Cyclooxygenase (COX) catalyzes the conversion of arachidonic acid (AA) into prostaglandin H2 (PGH2), which is subsequently converted by synthases and isomerases to the prostanoids PGE2, PGI2, PGF2α, and thromboxane A2 (TXA2). 27,34 The role of AA metabolites generated by COX in homeostasis of vascular tone and the clotting cascade has been well described. 38 Prostanoids also have roles in modulating renal blood flow, glomerular filtration rate, and salt and water excretion. 83 COX has 2 distinct membrane-anchored isoenzymes: COX-1 and COX-2. COX-1 is constitutively expressed in most normal tissues; COX-2 is highly induced by proinflammatory mediators in the setting of inflammation, injury, and pain (Figure 1 ). 81,83 In normal vessels, COX-1 is the predominant isoform, and its expression is constitutive in the endothelium but irregular in the vascular smooth muscles. 65,81,95,100,116,117 In contrast, COX-2 is not expressed in the majority of normal endothelial or vascular smooth muscle cells; however, it is rapidly induced with vessel trauma or inflammation. 27,65,95,116,117 Consistent with its role in inflammation, the 5′ flanking region of the COX-2 gene contains transcription factor binding sites for nuclear factor–kappa B, nuclear factor–interleukin 6, CCAAT/enhancer-binding protein, and a cyclic AMP response element. 95,100
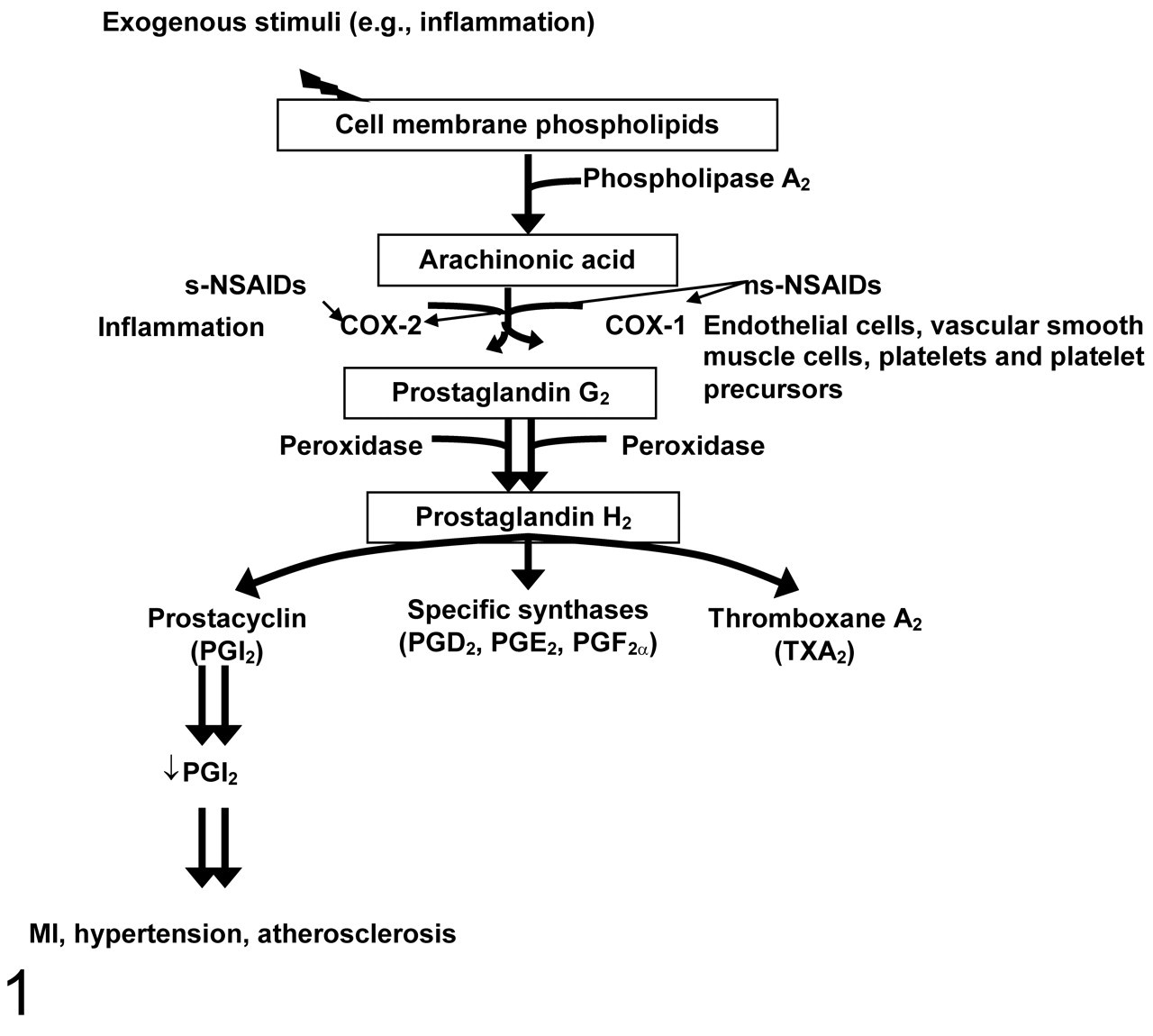
Prostaglandin synthesis: the role of prostaglandins in the cardiorenal system in health and disease and the effects of nonselective and selective nonsteroidal anti-inflammatory drugs (NSAIDs). Following an exogenous stimulus (eg, inflammation), cell membrane phospholipid is liberated to arachidonic acid by phospholipase A2. Two cyclooxygenases, COX-1 and COX-2, catalyze the conversion of arachidonic acid into various prostaglandins. COX-1 is expressed in endothelial cells, platelets, platelet precursors, and vascular smooth muscle cell, whereas COX-2 expression is upregulated during inflammatory or neoplastic conditions. Nonselective NSAIDs (eg, carprofen, etodolac, flunixin meglumine, ketoprofen, indomethacin, meloxicam, and phenylbutazone) inhibit COX-1 and COX-2; selective NSAIDs (eg, celecoxib, deracoxib, lumiracoxib, parecoxib, robenacoxib, rofecoxib, valdecoxib) spare COX-1 and inhibit only COX-2.
Numerous inhibitors of COX activity are presently on the market or have been withdrawn from the market and include conventional nonselective nonsteroidal anti-inflammatory drugs (ns-NSAIDs) and selective NSAIDs (s-NSAIDs). At therapeutic doses, ns-NSAIDs inhibit COX-1 and COX-2 (Figure 1), whereas s-NSAIDs favor COX-2 inhibition. 67 Examples of such ns-NSAIDs include acetylsalicylic acid (ASA; ie, aspirin), naproxen, ibuprofen, diclofenac, and phenybutazone. In contrast, s-NSAIDs—for example, robenacoxib (Onsior, Novartis Animal Health, Greensboro, NC), lumiracoxib (Prexige, Novartis Pharmaceuticals, Basel, Switzerland), celecoxib (Celebrex, Pfizer, New York, NY), rofecoxib (Vioxx, Merck & Co, Whitehouse Station, NJ), as well as others—provide anti-inflammatory effects with less of the connected COX-1-mediated toxicities, as assessed by studies of gastrointestinal mucosal integrity and renal injury. 83,84
An indicator of the COX-1 versus COX-2 activity at therapeutic doses is the COX-1:COX-2 half-maximal inhibitory concentration (IC50) ratio in human blood—that is, the concentration of the drug that inhibits 50% of the COX-1 activity in blood to that which inhibits COX-2 in the blood. This ratio is a good indicator of enzyme specificity; numbers greater than 1 are more COX-2 specific and those less than 1 are more COX-1 specific. There is wide variation between reports in the exact values, but the fold differences in COX selectivity of drugs tested have fairly good correlations between studies. Cryer and Feldman 23 reported that the ratio was 0.32 for ASA, whereas it was 0.60 for ibuprofen, 1.14 for naproxen, and 24.00 for NS-398. A report by Chan et al 19 indicated that the ratio in human blood was 36.0 for rofecoxib, 6.6 for celecoxib, and 0.4 for diclofenac.
The anti-inflammatory benefits of ns-NSAIDs are primarily derived from COX-2 inhibition. Inhibition of COX-1 by ns-NSAIDs causes disruptions in the microcirculation with the potential for gastrointestinal and renal ischemic injury. 62,81 However, the potential for untoward cardiovascular events associated with COX-2 inhibition, particularly in individuals with underlying cardiovascular disease, has been addressed in the last few years. 67 Preclinical studies have failed to identify these risks in a variety of animal species. Although a variety of experimental models have assessed the effects of s-NSAIDs and ns-NSAIDs, the outcomes have been variable. In this review, we discuss the role of COX in cardiovascular homeostasis and pathophysiology and examine the comparative differences across species. Additionally, we discuss effects associated with NSAIDs' pharmaceutical inhibition on the cardiovascular system.
COX-1 and COX-2 in Homeostasis in Health and Injury
The role of prostanoids in homeostasis and vascular tone has been well established. The effects of prostanoids are often opposing, which is important for maintaining a balance between vasoconstriction and vasodilation and between antithrombotic and prothrombotic events. Much of this balance is mediated through PGI2 and platelet-derived TXA2 and, to a lesser extent, PGE2. TXA2 is largely platelet derived (to a lesser extent, monocyte derived) and is the predominant AA metabolite in platelets. 87 Because TXA2 is platelet derived, it is primarily COX-1 dependent. Platelets express only COX-1 in humans and preclinical species; platelets have neither baseline nor induced expression of COX-2. 60,117 TXA2 causes local vasoconstriction, platelet aggregation, and smooth muscle cell proliferation. PGI2, which is primarily COX-2 derived, is a vasodilator with antithrombotic effects and is primarily expressed by endothelial and vascular smooth muscle cells. 27,65,100,116 PGI2 is essential for balancing the prothrombotic and vasoconstrictive effects of TXA2. 38,61 The exact source of baseline PGI2 is uncertain because there is little to no COX-2 expression in normal human endothelium. 65 However, COX-2 induction in damaged or inflamed endothelial cells promotes production of prostacyclin, which tempers prothrombotic events at those sites. Although PGE2 can cause vasodilation and modulate the effects of TXA2 on platelet aggregation, it does not directly participate in homeostasis.
Although a balance exists between TXA2 and PGI2 to control thrombosis, studies of PGI2 receptor–deficient (Ptgir –/–) and TXA2-overexpressing mice (B6;129Sv-Tbxa2Y385F ) have demonstrated that shifting the balance between these factors does not result in increased platelet aggregation or thrombosis under normal physiological conditions. 21,78 It is therefore likely that other homeostatic mechanisms play a role on clotting under normal conditions. Such molecules include nitric oxide, carbon monoxide, and CD39, to name a few, which play an important role not only in endothelial protection but also in prothrombotic and antithrombotic events and vascular tone. 72
With injury, the importance of a balance between PGI2 and TXA2 to modulate platelet aggregation becomes apparent. Vascular injury upregulates COX-2 activity and, therefore, local PGI2 production. If PGI2 receptor–deficient mice are subjected to vascular injury, they become significantly more prone to thrombotic events, as compared to wild-type (WT) controls. 21,78 Similar to PGI2 receptor–deficient mice, mice overexpressing the TXA2 receptor display increased neointimal hyperplasia, intimal cell proliferation, and platelet activation after mechanical artery injury. 21 The data indicate that the importance of a balance between PGI2 and TXA2 is essential, particularly with vascular trauma, although redundant controls via other molecules may predominate under normal physiological conditions.
Seta et al 93 utilized a global COX-2-knockdown approach (Ptgs2) in an effort to mimic the effect of COX-2 inhibition by s-NSAIDs. Their study revealed that mice with marked (but not complete) COX-2 inhibition had greater platelet aggregation along vessel walls after FeCL3-induced vascular injury, as compared to WT littermates. Ex vivo platelet activation studies have demonstrated no significant difference in platelet activation by thrombin or TXA2 production between the COX-2 knockdown and WT littermates. Thus, the increased platelet aggregation was likely attributable to COX-2-related changes in the vascular wall physiology. In addition, these mice had no alterations in the mean arterial pressure, thereby suggesting that under normal conditions, reductions in COX-2 do not notably alter blood pressure in mice. 93
PGE2, as derived from COX-1 and COX-2, promotes vasodilatation and promotes the thrombogenic potential of TXA2, although it is not by itself thrombogenic. 43 The receptors for PGE2, EP1–4, are G protein coupled. These receptors have differing signaling cascades. 28 Interestingly, high concentrations of PGE2 inhibit platelet aggregation by activating the PGI2 receptor (IP) nonspecifically. However, low concentrations of PGE2 potentiate the activation of partially activated platelets, thus increasing platelet aggregation. This potentiation is primarily through the activation of the PGE2 receptor EP3. 43
Effects of Pharmaceutical Inhibition of COX-1 and COX-2 on Homeostasis
The COX-1-inhibitory effects of NSAIDs have been associated with decreased platelet aggregation; these effects have been extensively studied. 17 The effects of COX-2 inhibitors, however, have been mixed and often contradictory; in animal studies, this may in part be due to variations in study design, genetic backgrounds, drug exposure, and the environment. In addition, under normal physiological conditions, alterations in platelet aggregation would not likely be identified with COX-2 inhibition, as demonstrated in the studies of PGI2 receptor–knockout and TXA2-overexpressing mice. Thus, in studies based on normal animal models, COX-2 inhibition would not necessarily cause significant reductions in PGI2 or TXA2 levels in vascular tissues; significant COX-2 inhibition would also not presumably alter platelet aggregation ex vivo. In healthy human volunteers, s-NSAIDs such as celecoxib have had no effect on the antithrombotic effect of ASA. 40,112
COX-1 and COX-2 in Cardiac Tissues in Health and Injury
Consistent with its expression in the peripheral vasculature, COX-1 in the heart of adult humans and animals is in endothelial cells and vascular smooth muscle. 65,97,116,117 In dogs, COX-1 expression in the heart has been localized to the fibrous connective tissue of the tricuspid valve and chordae tendinae. 97 In rats, COX-1 has been identified in the endothelial cells of the heart but not cardiomyocytes (N.K.K., unpublished data). Unlike COX-1, COX-2 has limited expression within the normal heart. COX-2 has been demonstrated by immunohistochemistry to have restricted and multifocal expression in aortic vascular endothelial cells in the dog and the rat; no expression has been identified in the human aorta. 59 In the cardiomyocytes of normal adult humans, dogs, and rodents, COX-2 expression is rarely evident. 116 In one study, reverse transcription polymerase chain reaction (RT-PCR) of normal mouse and rat hearts revealed scant COX-2 and high COX-1 expression, consistent with findings in the human and dog. 103 Although the authors in this study identified extensive COX-1 and COX-2 immunohistochemical staining in the cardiomyocytes of rats and mice, the data were somewhat inconsistent with their RT-PCR data, which may be explained by their use of a rabbit polyclonal antibody to stain the heart sections: In the experience of R.S.S., rabbit polyclonal antibodies extensively stain the myocardium, regardless of antigen and are thus thought to be unreliable for evaluating rodent hearts. A recent review article discusses COX-2-associated kinase cascades in the heart, with an excellent discussion of the various signaling cascades and posttranscriptional modifications related to COX-2 signaling in the heart. 100
Genetically engineered mice have allowed a better understanding of the role of prostanoids and COX in the heart. The evaluation of cardiac function and histology using mice null for COX-2 has had limited value, given that COX-2-null mice (Ptgs2) often die at birth and rarely survive out to 6 months of age. 31 Furthermore, the surviving mice develop myocardial fibrosis and have significantly underdeveloped kidneys and poor renal function; as such, discerning whether the cardiac change is primary or secondary is challenging. The use of site-specific and inducible gene-modulating technology has promoted the better understanding of organ- and time-specific changes in gene expression, particularly for genes whose mutation results in early death. Wang et al 108 recently published a study on the role of COX-2 in the adult mouse heart using a cardiomyocyte-specific inducible deletion of Ptgs2. Two weeks after the cardiomyocyte-specific deletion of COX-2 in postweanling animals, the mice had reduced systolic ventricular function, decreased heart rate, increased susceptibility to induced ventricular arrhythmias, and decreased exercise tolerance. These mice had partially elucidated the role of COX-2 in myocardial pathology associated with increased afterload. 108 When afterload was increased through aortic banding, the null mice had greater cardiomyocyte hypertrophy and fibrosis than that of their WT littermates. Interestingly, connexin 43, an important gap junction protein, was reduced in the COX-2-deficient cardiomyocytes as compared to that of littermates, and fibroblasts obtained from these hearts had greater expression of atrial natriuretic peptide, B-type natriuretic peptide, and matrix metalloproteinase 9.
In contrast, genetically engineered mice with reductions (knockdown) in COX-2 expression to levels interpreted to be similar to that induced by s-NSAIDs had no alterations in mean arterial pressure, heart rate, diastolic, or systolic pressure, as compared to WT littermates. 93 These 2 studies 93,108 indicated that COX-2 is essential for normal cardiac function but that reductions in COX-2 expression (as compared to deletion of COX-2 expression) do not significantly affect myocardial function. Mice that specifically overexpress COX-2 in the heart have demonstrated no differences in cardiac function, weight, or histology under normal conditions. 55
Although COX-2 is minimally expressed in the cardiovascular system constitutively, it is readily observed in damaged cardiac tissues in humans and animal models of myocardial disease. Immunohistochemical evaluation of human hearts for COX-1 and COX-2 post–myocardial infarction (post-MI) demonstrated that COX-1 and COX-2 expression was present in inflammatory cells, myofibroblasts, and the capillaries of granulation tissue and fibrosis. In human patients with cardiac dysfunction (ie, heart failure) and MI, abundant expression of COX-2 mRNA and protein has been detected in viable cardiomyocytes adjacent to the infarcted region as well as in inflammatory cells and vessels associated with regions of damage and scar formation. 2,116 As anticipated, COX-2 expression has been identified in the hearts of patients with septicemia and inflammatory heart diseases (myocarditis), as well as injury. 24,100 In a rat model of chronic MI and heart failure, strong COX-2 protein expression was observed by immunohistochemistry in the myocytes, endocardium, vascular endothelial cells, and macrophages in the viable tissue adjacent to the infarcted zone, as compared to nominal expression noted in control rats. 90 These data indicate that rodent models and human COX-2 responses in MI are similar.
Interestingly, prostaglandins may play a role in myocardial hypertrophy. Overexpression of the PGE2 receptor gene Ptger3 in mice with the alpha-myosin heavy chain promoter resulted in marked myocardial hypertrophy with increased expression of transforming growth factor β, atrial natriuretic peptide, cardiac ankyrin repeat protein, and connective tissue growth factor with increased interstitial fibrosis in the transgenic mice, as compared to the WT littermates. 74 These mice had marked increases in end diastolic and systolic volumes at 5 to 6 weeks of age, as compared to the WT littermates. In addition, the left ventricular ejection fraction was markedly decreased in the transgenic mice, suggesting reductions in contractility. Hypertrophic effects of Ptger3 overexpression in these mice was thought to be mediated by increased activation of the calcineurin signaling pathway. Thus, PGE2 may be a factor in the development myocardial hypertrophy. However, deletion of Ptger3 in the heart would be required to identify if the reduction of PGE2 signaling could cause myocardial atrophy and dysfunction.
Effects of COX-1 and COX-2 Inhibition in Myocardial Ischemia and Infarction
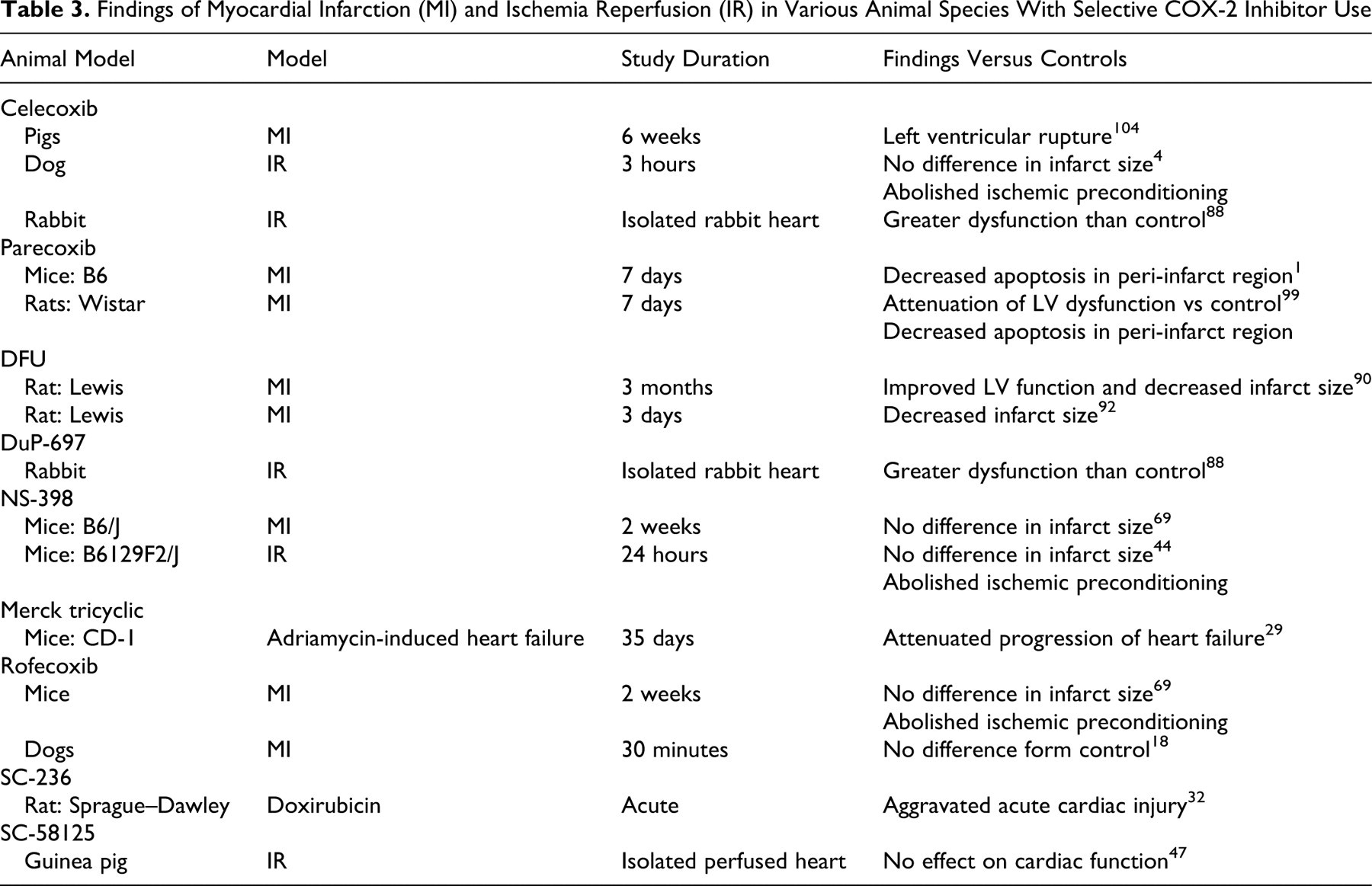
Animal models of the effects of COX inhibition in myocardial ischemia have been contradictory (Tables 1–3). Animal models of MI generally fall into 4 categories: MI, ischemia with reperfusion (IR), IR with ischemic preconditioning, and IR with postconditioning. For a detailed review of these effects, see Ferdinandy et al. 36 The majority of animal models evaluating the cardiac effects of COX inhibition have utilized either MI (complete coronary artery ligation) or IR (coronary artery ligation for several hours, followed by reperfusion) with or without ischemic preconditioning. Although reperfusion after ischemia is necessary for tissue survival, reperfusion results in the release of reactive oxygen species that may incite further cell death. The duration of the ischemic event before reperfusion affects the degree of tissue injury. 36 A few examples are included here, but a summary of the drugs, types of studies, and outcomes can be found in Tables 1–3.
Examples of Myocardial Infarction (MI) and Ischemia With Reperfusion (IR) in Various Animal Models With Acetylsalicylic Acid a

a Aspirin as cyclooxygenase (COX-1) inhibitor. Half-maximal inhibitory concentration (IC50) ratio: COX-2:COX-1, approximately 3.12. 23
Findings of Myocardial Infarction (MI) and Ischemia Reperfusion (IR) in Various Animal Models With Use of Nonsteroidal Anti-inflammatory Drugs
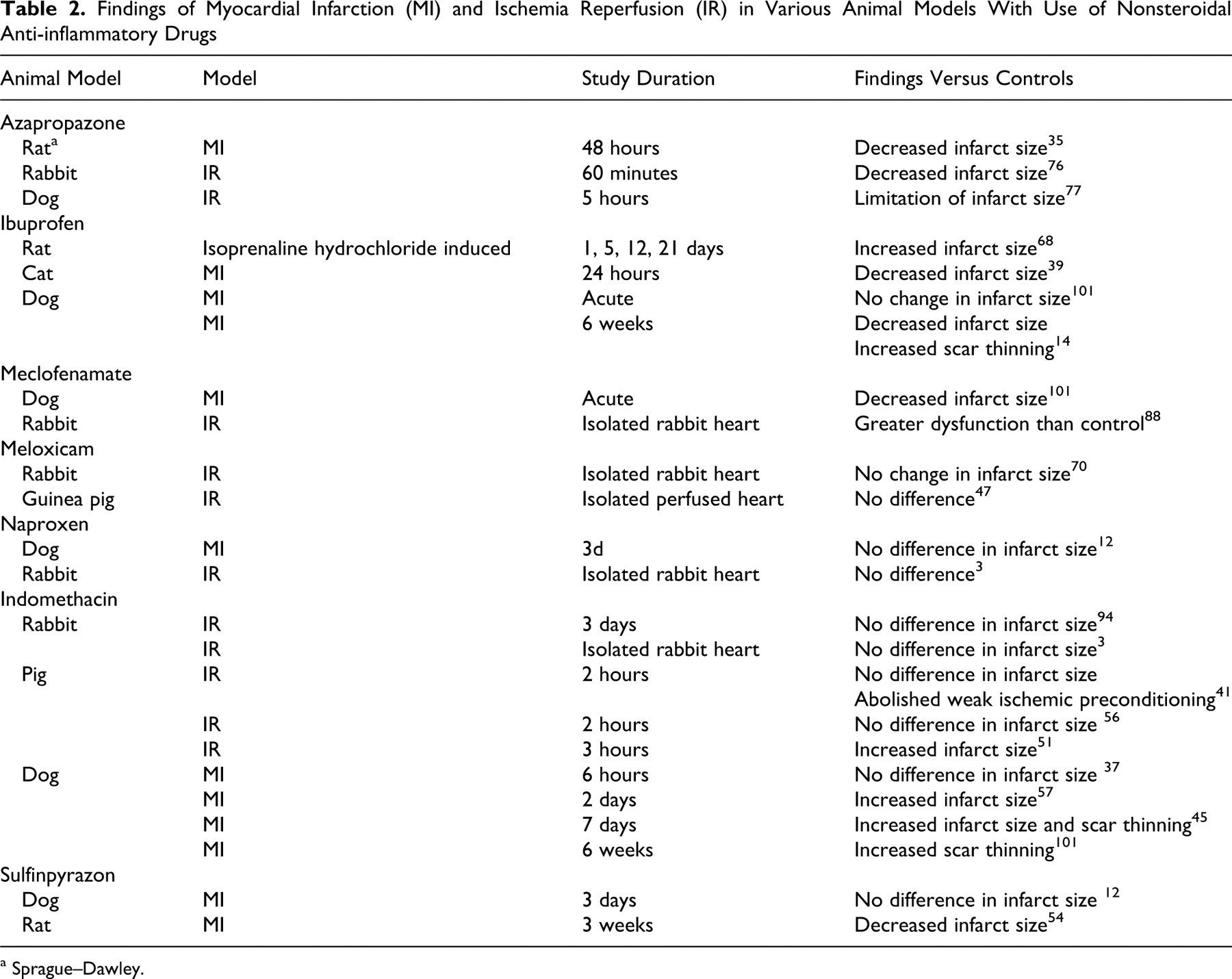
a Sprague–Dawley.
Findings of Myocardial Infarction (MI) and Ischemia Reperfusion (IR) in Various Animal Species With Selective COX-2 Inhibitor Use
Genetically engineered mice: IR and MI studies
Studies in which mice with COX-2 specifically overexpressed in cardiomyocytes (B6D2-Tg(MHC-Ptgs2)17Upme) were subjected to transient coronary artery occlusion (without preconditioning) followed by reperfusion demonstrated improved contractility during the reperfusion phase and decreased hypercontracture, infarct area, and lactate dehydrogenase release, as compared to the WT mice. 55 The magnitude of this effect was similar to that seen with ischemic preconditioning. In WT and COX-2 overexpressing mice, the PGE2, PGI2, and PGF2α increased but not TXA2. Only PGE2 was significantly higher in transgenic mice than in WT mice, suggesting that this molecule is important in the prevention of cell death during IR events. PGE2 receptor–overexpressing mice (Ptger3) subjected to IR also demonstrated significant protection against ischemia-associated creatine phosphokinase release and left ventricular contracture. 74 The authors hypothesized that this is related to blunting of ischemia-induced endogenous catecholamines by attenuating of the cAMP-mediated calcium influx into cardiomyocytes. However, induced MI in microsomal prostaglandin E2 synthase 1–null mice (Ptegs1) revealed similar effects on post-MI survival, infarct size, or creatine phosphokinase and cardiac troponin I levels, as compared to control animals, indicating that inflammation-induced PGE2 may not significantly contribute to the outcome in MI. 114
COX inhibition in MI studies
COX inhibition in acute MI studies can be divided into short- and long-term findings, with most studies having a duration of fewer than 7 days. Treatment with an s-NSAID—DFU, or 5,5-dimethyl-3-(3-fluoropheny11)-4-(4-methyl-sulphonyl-2(5H)-fluranone—in Lewis rats for 3 days and 3 months after coronary artery ligation revealed a smaller infarct than that of controls, with better left ventricular function than that of vehicle-treated animals. 90,92 Wistar rats treated with the s-NSAID parecoxib (Pfizer, New York, NY) for 5 days, beginning 2 days post–coronary artery occlusion, had increased wall thickness at the site of the infarct and improved cardiac function when compared to vehicle-treated animals. 99 An interesting study on Wistar rats examined the effect of COX-2 inhibition after prolonged heart failure. Specifically, these rats underwent coronary artery ligation to induce ischemic congestive heart failure. They were treated 1 year after the ligation with parecoxib for 7 days, a COX-2 inhibitor, and demonstrated increased myocardial function in the peri-infarct area. 1
Studies of Landrace pigs treated with parecoxib for 7 days after MI demonstrated only decreased apoptosis around the infarct, as compared to vehicle control animals. Compared to vehicle-treated animals, Landrace pigs treated with ASA for 7 days before MI had no difference in infarct size or heart function. Those treated with a derivative of ASA, NCX 4016, did have reduced infarct size, when compared to ASA- or vehicle-treated pigs. 107 Landrace pigs treated for 6 weeks with celecoxib (plus clopidrogel and sotalol) after induction of MI had increased risk of mortality from left ventricular rupture at the site of the infarcts. The area of infarction had decreased left ventricular wall thickness and decreased collagen within the infarcts, as well as decreased cardiovascular function. 104
Dogs treated with ASA, rofecoxib, or meclofenamate in acute MI studies revealed reductions in the infarct size, 18,101 but long-term studies with ASA (6 weeks post-MI) revealed no difference in infarct size from that of control animals. 14 Compared to vehicle-treated animals, dogs treated with ibuprofen for up to 24 hours or with ASA for up to 3 hours after MI had increased scar thinning 6 weeks after MI. 14
In summary, these studies suggest that COX-1 inhibition may have little impact on short- and long-term effects of MI. COX-2 inhibition may acutely reduce infarct size and inflammation at the site of MI. The reduction in local inflammation may have short-term value but long-term consequences in tissue breakdown and scar formation. Although the findings after long-term inhibition of COX-2 after infarction are variable, a trend in these studies indicates decreased collagen density and normal wall remodeling at the infarct site, which may result in scar thinning and adversely affect heart function.
COX inhibition in MI studies IR studies
Dogs, rats, and mice who undergo several cycles of ischemic preconditioning (4 to 5 minutes of ischemia followed by 4 to 5 minutes reperfusion, repeated 4 to 5 times) have reduced myocardial injury when this preconditioning is followed by prolonged IR. 4,36,44 The effect of ischemic preconditioning is lost if s-NSAIDs such as celecoxib and NS-398 are administered before surgery, whereas the protective effect of preconditioning on the myocardium is not lost with pretreatment with ASA. 4,44 These studies demonstrate the importance of COX-2, not COX-1, in ischemic preconditioning–associated myocardial protection. Studies are also limited on the effect of COX inhibition in IR without preconditioning. Many of these studies focused on the early and immediate effects of COX inhibition in reperfusion, rather than the long-term consequences in remodeling. A study on dogs treated with an s-NSAID, rofecoxib, before 3 hours of cardiac ischemia followed by 30 minutes of reperfusion showed no detectable differences in creatine phosphokinase–MB or troponin I, as compared to controls, 18 suggesting that acute injury is not altered in IR injury with COX inhibition. A longer-term study in rats exposed to 8 hours of cardiac ischemia followed by 2 weeks of reperfusion and ASA therapy revealed no differences in infarct size, septal, or left ventricular wall thickness, as compared to vehicle-treated animals. 5
In summary, COX-2 is an important mediator in the protective effect of preconditioning in IR studies. However, neither COX-1 nor COX-2 inhibition appears to significantly affect the outcome in IR studies of several animal species.
COX-1 and COX-2 in Atherosclerosis
Atherosclerosis is one of the most important contributors to cardiovascular disease in humans and nonhuman primates. It is a complex inflammatory disease of medium- and large-sized vessels that results in intimal accumulation of lipids, macrophages, matrix, mineral, and other inflammatory infiltrates, as well as smooth muscle cell proliferation. 75 It is considered to be a predominantly inflammatory disease, and during initiation of atherosclerotic plaques, inflammatory cells, along with lipids, infiltrate the vascular wall. 22,86 The subsequent cytokine, cell signaling, and enzyme production promote plaque growth (refer to reviews by Moubayed et al 75 and Cipollone et al 22 ).
Prostaglandins promote atherosclerosis by altering the inflammatory response and expression of matrix metalloproteinases, as well as by acting as a mitogen for various cell types, including vascular smooth muscle cells. Matrix metalloproteinase 2 and 9 upregulation in plaques has been demonstrated to be in part mediated by COX-2- and COX-1-derived PGE2 22 and thus likely contribute to plaque rupture. TXA2 is clearly an important mediator of plaque development and progression. 25 The source of TXA2 increase in atherosclerosis is primarily derived from COX-1 in platelets. 9 Increases in PGI2 are the result of both COX isoforms in the endothelium, although much of the PGI2 increases in atherosclerosis have been attributed to COX-2 upregulation at the site of the plaque. 8 In the region of atherosclerotic plaques, COX-2 expression is present in endothelial cells, vascular smooth muscle cells, and macrophages. 9 As in humans, mouse and rabbit models of atherosclerosis have COX-2 expression in fatty streaks and atherosclerotic vessels. 8,15,52,98,113 These data indicate that in animal models of atherosclerosis, the expression of COX isoforms is similar to that of humans.
Mammalian models of atherosclerosis have shed light on the pathogenesis of this disease and the roles of COX-1 and COX-2 on the development and progression of atherosclerotic plaques. Mammalian species other than primates do not spontaneously develop atherosclerosis under normal conditions; in primates, the lesion is generally associated with aging. This fact certainly presents limitations in the evaluation of cardiovascular risks associated with atherosclerosis in the typical preclinical species and in the relatively young primates used in such preclinical studies. Animal models utilized in the study of atherosclerosis and its development include the PGI2 receptor–deficient mice (Ptgir –/–), the apolipoprotein E–deficient mouse (Apoe –/–), the LDL receptor–deficient mouse (Ldlr –/–), and the ApoE*3-Leiden transgenic mouse (Tg(APOE*3,APOC1)2Lmh), as well as the high-fat-diet-induced atherosclerosis in hamsters and Watanabe rabbits. 33,64,66,105,115 For an excellent review of mouse models of atherosclerosis, see Zadelaar et al. 115
The importance of prostaglandins in the development of atherosclerosis has been demonstrated with several animal models of atherosclerosis. Studies by Kobayashi et al 64 using mice mutant for Apoe and either the PGI receptor (Ptgir) or the TXA2 receptor (Tbxa2r) has helped to elucidate the importance of prostacyclin and thromboxane in the development of atherosclerosis. In Apoe–/– Ptgir–/– mice, atherogenesis was accelerated when compared to that of Apoe–/– mice. In contrast, in Apoe –/– Tbxa2r –/– mice, atherogenesis was delayed as compared to that of Apoe–/– mice. 64 These data indicate that PGI2 is protective against the atherosclerosis whereas TXA2 is important in the initiation and progression of atherosclerosis. 33,64 This finding is in part due to modulation of platelet aggregation and leukocyte–endothelial cell interactions through alterations in the adhesion molecules ICAM-1 and PECAM-1. Studies of Apoe–/– Ptgs1–/– (COX-1) mice fed a 1% cholesterol diet for 8 weeks showed decreased atherosclerotic lesion development, as compared to control mice. 73 These data implicate COX-1 in the early pathogenesis of atherosclerosis. Interestingly, platelet adhesion to vessel walls was not notably different between double mutants and controls under normal conditions; however, reductions in platelet adhesion were evident after vascular ligation injury in the Apoe–/– Ptgs1–/– mice, as compared to controls, which suggests that COX-1 is also important in promoting platelet adhesion to damaged vessel walls and is likely mediated through TXA2 production. Studies of hamsters have also demonstrated that PGI2 imparts atheroprotective properties and that analogs of PGI2 retard atherogenesis, which is consistent with genetically engineered mouse studies. 66
Effects of Pharmaceutical Inhibition of COX-1 and COX-2 on Atherogenesis
Studies of atherosclerotic mouse models have been divided into evaluation of initiation and evaluation of progression. Studies in Apoe-null mice have demonstrated that COX-1 inhibition appears to consistently modify the initiation and/or progression of atherosclerosis. 9,26 Further evidence that TXA2 and COX-1 are important in atherogenesis is from Ldlr-deletion mice treated with TXA2 receptor blockers and COX-1 blockers. These mice had reductions in atherogenesis as compared to that of untreated animals. 25 In cholesterol-fed rabbits, an ns-NSAID, indomethacin, reduced the progression of atherosclerotic lesions and improved endothelium-mediated vascular responses. 96 COX-2 inhibition with rofecoxib promoted early atherosclerotic lesion formation in Ldlr-deficient mice, and COX-2 inhibition with Merck Frosst tricyclic increased both early atherosclerotic lesion area and atherosclerotic plaque destabilization in Apoe-deficient mice. 16,89 In Apoe-deficient mice, COX-2 inhibition, using SC-236, demonstrated that despite selective reduction in prostacyclin generation, there is no effect on lesion development or platelet interactions with the vessel wall. 9 Other studies demonstrated that chronic administration of s-NSAIDs (celecoxib and Merck Frosst tricyclic) do not influence or have an effect on the composition of advanced atherosclerotic lesions in the same mouse strain. 7,80,82 Ultimately, the data from animal models suggest that although COX-2 is highly expressed in atherosclerotic plaques, it likely plays a lesser role than that of COX-1 in lesion development and progression.
Blood Pressure and COX Expression
COX-1 and COX-2 have effects on localized vascular bed pressure, as previously described. Systemic blood pressure is primarily maintained by the renin-angiotensin-aldosterone system, and hypertension is often due to abnormal renal vascular control homeostasis (for recent review, see Radi 83 ). This homeostatic mechanism has been well described and is reviewed in detail here.
Studies in COX-1-null mice under conditions of reduced sodium intake revealed a mild but significant reduction in blood pressure over that of control mice. 6 The data study indicate that COX-1 is important in indirectly maintaining blood pressure by maintaining sodium retention. In addition, blood pressure has a circadian pattern, with reductions in blood pressure during sleep. 58 COX-1-null mice have impaired reductions in blood pressure during sleep, 58 indicating that COX-1 may be important in maintaining normal circadian rhythms in blood pressure. COX-2 knockdown mice have no notable alterations in systemic blood pressure as compared to that of control mice. 93 These data indicate that, under normal conditions, the effects of COX on systemic blood pressure are likely primarily mediated through its actions on renal sodium retention and excretion.
Effects of Pharmaceutical Inhibition of COX-1 and COX-2 on Coronary Blood Flow and Blood Pressure
The effects of COX inhibition on local blood flow in the coronary artery have been evaluated in dogs. The models have evaluated the ability of COX inhibitors to modulate coronary vasodilatation and blood flow induced by AA and acetylcholine (ACH). In these studies, AA-induced coronary blood flow is attenuated by pretreatment with ASA, nimesulide, naproxen, and celecoxib. 48,53 In contrast, ACH-induced coronary vasodilatation (nitric oxide–dependent pathway) in dogs is unaffected by pretreatment with naproxen and SC-58236, an experimental s-NSAID, and SC-58236 fails to significantly attenuate either AA- or ACH-induced vasodilatation. 42 These data suggest that the effects of AA, at least in the dog, are primarily COX-1 dependent.
The effect of COX inhibition on blood pressure in preclinical species has been variable. Studies in normal animals indicate that ns-NSAIDs and/or s-NSAIDs have little effect on the systemic arterial blood pressure. 10,11,13,71 In normotensive human patients, blood pressure and renal function are not significantly altered by short-term treatment with standard doses of celecoxib (200 mg twice a day). 30 Although blood pressure studies of dogs appear to be consistent, there is reported variation between rat strains in the effects of COX-2 inhibition on blood pressure. A study comparing the effects of diclofenac, celecoxib, and rofecoxib in Dahl salt–sensitive rats found that after 8 weeks of treatment, celecoxib slightly but significantly reduced systemic blood pressure (about 4%) as compared to controls and that diclofenac and rofecoxib minimally but significantly increased systemic blood pressure (about 1%). 49 In addition, COX-2 inhibition in Hannover Sprague–Dawley rats has no effect on systemic blood pressure. 106 In contrast, COX-2 inhibitors have been found to increase blood pressure in normotensive and hypertensive Wistar rats. 50,79 In a model of renovascular hypertension using Wistar–Kyoto or Sprague–Dawley rats, COX-2 inhibition had no significant effect on blood pressure. 46,85 Many of the differences in the decreased cardiovascular risk with celecoxib, as compared to rofecoxib, may be attributable to its decreased COX-2 selectivity. If this hypothesis is correct, it suggests that some COX-1 inhibition, along with COX-2, may be important to prevent thrombotic events in at-risk populations. 67
The effects of COX inhibition on aged humans and animals may be of greater clinical significance. Studies of the aortic endothelium from aged rats demonstrated increased Ptsg1 (COX-1) and Ptsg2 (COX-2) gene expression. 63,102 These studies also found that aortic endothelial cells from hypertensive rats had increased COX-1 gene expression. The authors suggested that increased COX-1 expression may explain why, in aging animal, endothelial cells tend to secrete more COX-derived vasoconstrictive prostanoids than those of younger animals. 63,102 Studies in aged animals are uncommon; thus, studies of the effects of COX inhibition in aged animals are limited. Interestingly, one study found that in aged humans (66 ± 3 years) with coronary artery disease, COX-2 inhibition was associated with improved coronary artery endothelium-dependent vasodilation. 20
There is some indication that COX inhibition may attenuate the effects of some antihypertensive therapeutics. In a study of human patients taking antihypertensives, COX-2 inhibitors increased systolic and diastolic blood pressure but to different severities, depending on the drug (eg, the effects on blood pressure were greater in people treated with rofecoxib than with celecoxib). 109,110 For patients on ACE inhibitors, high doses of celecoxib had no statistically significant effects on blood pressure. 111
Conclusions
COX-1- and COX-2-derived prostaglandins are essential in normal cardiovascular physiology. Postmarketing withdrawal of certain COX-2 inhibitors, based on concerns of cardiovascular complications, has raised the question why no adverse cardiovascular findings were identified in preclinical studies before their use in the general population. A thorough review of preclinical toxicology studies, which included hemodynamic and detailed morphologic assessments, revealed no treatment-related adverse findings in cardiovascular function or histopathology for a broad range of doses of celecoxib, valdecoxib (Bextra, Pfizer, New York, NY), rofecoxib, and meloxicam after repeated dosing in multiple rodent and nonrodent species. The 2-year chronic toxicity studies indicated that standard preclinical toxicology studies in multiple species at broad dose ranges and up to 2 years in duration did not predict the adverse cardiovascular safety events identified in long-term clinical studies.
Animal models of cardiovascular diseases and genetically engineered mice have provided some indirect information in understanding potential functions of COX-mediated prostaglandins in homeostasis and disease. The exact mechanisms by which NSAIDs are associated with cardiovascular risk in humans remain somewhat elusive; the understanding of this risk is exacerbated by notable species and strain variation in the effects of COX inhibition in animal models of cardiovascular disease. Although disease models may be considered in assessment of the overall safety profile of a new therapeutic candidate, species and strain variations in response may not be predictive of human risk.
Footnotes
A time of publication, both Z. A. Radi and N. K. Khan were employed by Pfizer.
The authors declared that they received no financial support for their research and/or authorship of this article.