
Abstract
Lynch Syndrome is an autosomal dominant cancer predisposition syndrome caused by germline pathogenic variants or epimutation in one of the DNA mismatch repair genes. De novo pathogenic variants in mismatch repair genes have been described as a rare event in Lynch Syndrome (1-5%), although the prevalence of de novo pathogenic variants in Lynch Syndrome is probably underestimated. The de novo pathogenic variant was identified in a 26-year-old woman diagnosed with an adenocarcinoma of the caecum with mismatch repair protein deficiency at immunohistochemistry and a synchronous neuroendocrine tumor of the appendix with normal expression of mismatch repair proteins. DNA testing revealed deletion of exon 6 of the MLH1 gene. It appeared to be a de novo event, as the deletion was not detected in the patient’s parents. The presence of a mosaicism in the patient was excluded and haplotype analysis demonstrated the paternal origin of the chromosome harboring the deletion. The de novo deletion probably originated either from a very early postzygotic or a single prezygotic mutational event, or from a gonadal mosaicism. In conclusion, the identification of de novo pathogenic variants is crucial to allow proper genetic counseling and appropriate management of the patient’s family.
Introduction
Lynch syndrome (LS) is a hereditary autosomal dominant disease characterized by an increased risk of developing many types of cancer, mainly colorectal (CRC) and endometrial cancer and less frequently ovarian, gastric, small bowel, hepatobiliary tract, pancreatic, urinary tract, skin and brain cancers, 1 usually occurring at a young age.
LS is defined by the presence of a germline heterozygous pathogenic variant or epimutation in one of the DNA mismatch repair (MMR) genes (MLH1, MSH2, MSH6 or PMS2). These genes encode proteins involved in DNA repair mechanisms. Loss of expression of MMR proteins is indicative of loss of MMR activity that leads to accumulation of DNA replication errors, especially in repetitive sequences, inducing microsatellite instability in the tumor tissue.
In individuals carrying a germline pathogenic variant in one of the MMR genes, the lifetime risk of cancer depends on which MMR gene is mutated and on the sex of the individual. According to the Prospective Lynch Syndrome Database (PLSD),2,3 MLH1 and MSH2 are high-penetrance genes, with a cumulative cancer risk by age 70 of 64–78%. Of note, MSH6 pathogenic variants predispose mainly women to cancer (cumulative cancer risk by age 70 of 62% in women versus 28% in men) while PMS2 pathogenic variants confer an overall cumulative cancer risk similar to the general population risk (22% versus 20% by age 74).
The risk for CRC associated with MLH1 and MHS2 pathogenic variants is significantly higher than the risk associated with MSH6 or PMS2 pathogenic variants. In particular, the lifetime risk of CRC is 44–53% for MLH1, 42–46% for MSH2, 12–20% for MSH6, and 3% for PMS2, depending on gender.
The highest risk of developing endometrial cancer occurs in patients with MSH2 and MSH6 pathogenic variants (46% and 41% by age 70, respectively), followed by MLH1 (35%), and PMS2 (13%).
Most of the time, LS is inherited. De novo pathogenic variants (PVs) in MMR genes have been described as rare events. Results from a study on 261 mutation carriers revealed that de novo PVs in MMR genes account for 1-5% of LS patients. 4 To date, a total of 14 de novo PVs in MMR genes have been reported in LS patients: 5 six in MLH1, six in MSH2, and two in MSH6. The majority were single nucleotide variants (SNVs) (n=11) and three were deletions. In addition, three de novo mosaicisms have been reported, probably due to variants arising in later post-fecundation phases.6-8 However, it is likely that the frequency of LS resulting from de novo PVs is underestimated, and the identification of de novo PVs in LS is essential to allow appropriate management of the patient itself, excluding the possibility of a sporadic dMMR tumor, and of its family, avoiding unnecessary screening.
The aim of the study is to describe a deletion in the MLH1 gene, found for the first time as a de novo variant.
Clinical presentation
The proband was a 26-year-old pregnant woman presenting with persistent abdominal pain, anemia and melena. Inflammatory bowel disease was suspected and Caesarean section was planned, also because of fetal development delay and distress. On post-partum colonoscopy an ulcerated, bleeding mass was found in the caecum and a right hemicolectomy was performed. The histology report indicated a poorly differentiated adenocarcinoma of the caecum, T3 N1a (70 nodes analyzed), associated with a well-differentiated neuroendocrine tumor of the appendix. According to the regional recommendations on universal screening for LS, immunohistochemistry of MMR proteins was performed on the caecal adenocarcinoma and showed loss of expression of MLH1 and PMS2 (dMMR). On the contrary, the neuroendocrine tumor of the appendix shows proficient MMR expression (pMMR).
In the proband’s family, only one other cancer of the LS spectrum was known: the maternal grandmother had been treated for sigmoid cancer in 2017, at the age of 86 years (Figure 1). Of note, immunohistochemistry on the grandmother’s tumor tissue, performed at the time of diagnosis, did not reveal loss of MMR protein expression.
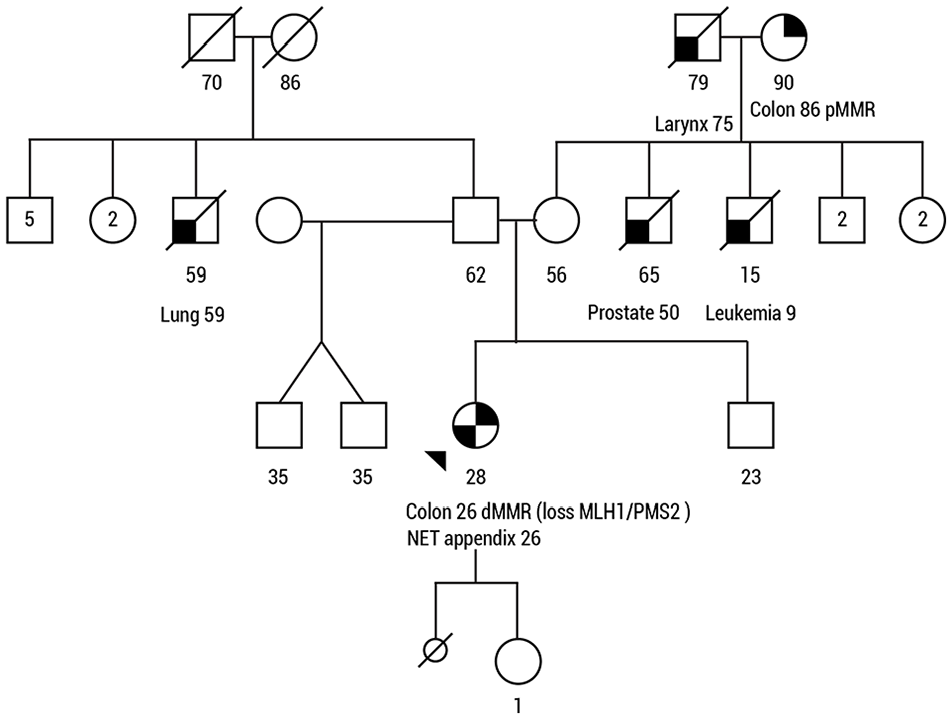
The patient’s family pedigree.
Given her age and the deficient MMR phenotype, the patient was referred for genetic counseling and testing.
The patient’s DNA extracted from blood was analyzed with a custom hybrid capture-based Next Generation Sequencing (NGS) panel named OncoPan. 9
NGS analysis of the MMR genes revealed deletion of the entire exon 6 of the MLH1 gene in heterozygosity in the patient’s DNA, confirmed by Multiplex ligation-dependent probe amplification (MLPA) analysis on a second independent aliquot of blood (Online Supplementary Figure 1S). The deletion is classified as pathogenic in the InSiGHT variant database since it introduces an early termination site and has been already reported several times. The HGVS nomenclature of the variant is the following: NM_000249.4:c.(453+1_454-1)_(545+1_546-1)del p.(Glu153Phefs*8). Analysis of the other MMR genes (MSH2, MSH6, PMS2 and EPCAM) revealed no other pathogenic variants.
To determine the parental branch to be investigated, MLPA analysis and digital Polymerase Chain Reaction (dPCR) on two independent aliquots of blood were carried out in the proband’s parents, but both tested negative for the deletion (Online Supplementary Figure 1S and Figure 2). Paternity was tested through haplotype analysis and the genetic fingerprint confirmed the parental status with convincing evidence (probability of paternity >99%) (Online Supplementary Figure 2S).
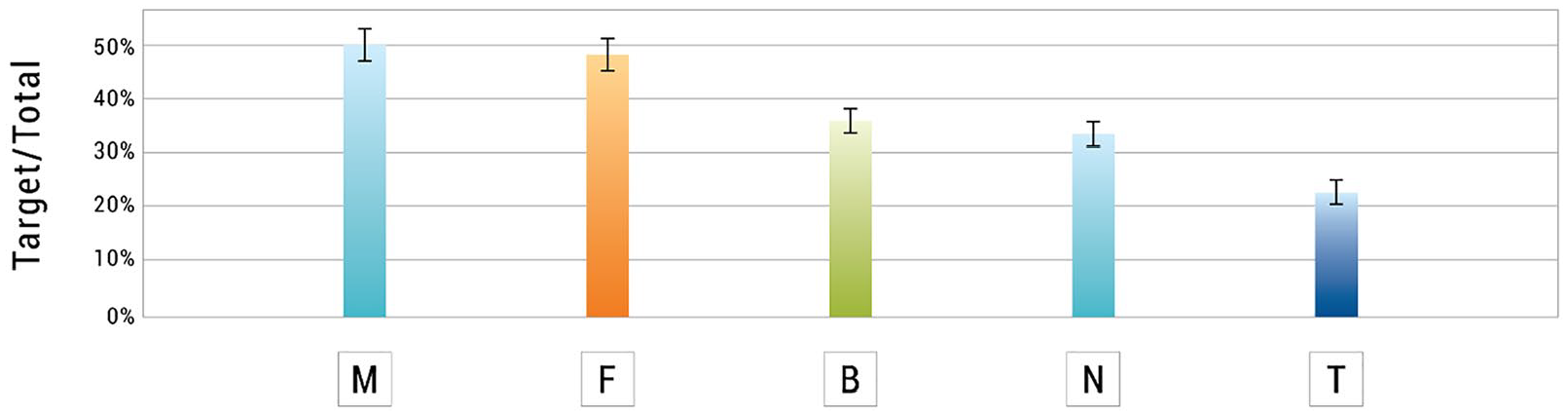
Digital PCR analysis: quantification of the presence of the exon 6 MLH1 deletion in the DNA extracted from the mother’s blood (M), father’s blood (F), patient’s blood (B), patient’s non-neoplastic tissue (N), patient’s tumor tissue (T).
Finally, the parental origin of the chromosome involved in the mutation was determined using the c.-93G>A, rs1800734 SNP in the MLH1 gene, previously detected in heterozygosity in the patient. The mother was homozygous A>A, while the father was heterozygous G>A. Long-range PCR of the region from exon 1 to exon 6 of the patient’s DNA was performed. As the mutation detected in the patient was the large deletion of the entire exon 6 of the MLH1 gene, amplification gave rise to the wild-type (WT) allele only, as the reverse primer in intron 6 was located inside the deletion. Nested PCR of exon 1, followed by Sanger sequencing, established that the WT allele carried an A at the rs1800734 locus and was of maternal origin. The mutated allele, carrying the pathogenic mutation, was thus of paternal origin.
As a gonadal paternal mosaicism was difficult to exclude, genetic testing was offered to the patient’s full brother and two paternal half-brothers, but they declined.
In order to rule out the possible presence of a mosaicism in the patient, in addition to the molecular analysis conducted by NGS on her blood, DNA extracted from healthy intestinal mucosa and tumor tissue from the caecum (cellularity ~80%) were also analyzed by MLPA and dPCR analysis (Supplementary Figures 1S and Figure 2).
The deletion of exon 6 was present at the same frequency of nearly 50%, thus in heterozygosity, both in the blood and the healthy mucosa, as assessed by MLPA and dPCR analysis, and this made it possible to exclude mosaicism in the patient.
The tumor tissue, on the other hand, had a higher frequency of the variant. In fact a copy number of 0.6, as detected by dPCR, indicates the presence of 30% of exon 6 in the tumor tissue and, as a consequence, of 70% of its deletion. Of note, all other exons of the gene were present in two copies as revealed by MLPA analysis.
To explain the increased frequency of exon 6 deletion in tumor tissue, despite the presence of two alleles of the gene, we can assume that the wild-type allele had been lost through LOH in the tumor, or in some of its cells, but subsequently the mutated allele carrying the deletion had undergone amplification. Otherwise a locus-restricted recombinational event, i.e., gene conversion, could have occurred, as this mechanism has been already observed in CRCs from MLH1 or MSH2 deletion carriers. 10
Discussion
The study reports an additional patient with a de novo deletion of exon 6 of the MLH1 gene. To the best of our knowledge, only four other articles in the literature showed a total of six de novo PVs in the MLH1 so far: four were point mutations4,11,12 and two were large deletions.4,13 De novo PVs in the MMR genes are reported to be a relatively rare event. 4 Likewise, the BRCA1/2 de novo PV rate is very low and was estimated to be 0.3%. 14 In contrast, other hereditary CRC predisposition syndromes have a higher de novo PV rate: for example, in APC-associated polyposis, de novo PVs account for up to 25% of patients, 15 in Peutz-Jeghers syndrome for 25-50% 16 and in juvenile polyposis syndrome for about 25%. 17 Indeed, the de novo PV rate has been shown to be higher in early-onset genetic diseases that impact on the reproductive capacity of individuals. 18
However, the prevalence of de novo PVs in LS is likely to have been underestimated due to the selection criteria for MMR genetic testing used in the past (Amsterdam criteria), which mainly took into account the family history of the proband.
Universal screening with microsatellite instability and/or immunohistochemistry staining for all patients with newly diagnosed CRC and endometrial cancer has been demonstrated to be more efficient in identifying LS patients, in particular in the setting of a negative family history. Negative family history can be explained not only by incomplete information about family history, reduced penetrance of MMR mutations, or non-paternity/non-maternity, but also by the presence of a de novo PV. The application of universal screening would improve de novo PV detection and help to understand the real prevalence of these mutations in LS. Besides, considering the high frequency of sporadic CRC, the possibility of phenocopies within an LS family should be considered (e.g., the grandmother’s pMMR CRC in the present family).
For these reasons, systematically testing parents of MMR mutation carriers, whenever possible, is essential to allow better management of the patient’s family. It is also important to exclude the possibility of a mosaicism. To date, only three patients with mosaicism in MMR genes have been described in the literature.6 -8
A mosaicism was excluded with a high level of confidence because the MLH1 deletion was present at the same frequency (nearly 50%) in her peripheral blood and healthy intestinal mucosa, and blood and intestinal mucosa do not originate from the same embryonic leaflet. Moreover, the negative mutation analysis result in the parents was consistent with the presence of a de novo PV in the patient originating either from a very early postzygotic or single prezygotic mutational event, or from a gonadal mosaicism.
As gonadal mosaicism in the parents is usually difficult to exclude, predictive genetic testing should be offered not only to the offspring but also the siblings of the patient.
Haplotype analysis demonstrated the paternal origin of the chromosome harboring the deletion. De novo PVs of paternal origin seem to be more frequent than those of maternal origin; 19 they are due to errors accumulated during the many DNA replication cycles required for spermatogenesis.18,19
Furthermore, like in previously reported patients, the present de novo PV was identified in a young woman diagnosed with CRC, confirming that patients with de novo PVs seem to develop cancer at an earlier age than carriers of inherited mutations. The earlier age at cancer diagnosis observed in patients with de novo PVs in the MMR genes compared to carriers of inherited mutations may be related to the co-occurrence of other de novo alterations in other parts of the genome, thus causing anticipation.
However, this observation may be due to an ascertainment bias. In fact, it is more likely to be able to confirm the presence of a de novo PV by testing both parents in LS patients with a young age at cancer diagnosis. This fact may contribute to the underdiagnosis of de novo PV in LS patients with a later age at cancer diagnosis.
Finally, despite NETs have been already reported in LS patients,20 -23 there is no evidence in the literature of a direct correlation between them. More importantly, the adenocarcinoma exhibited MMR protein deficiency at immunohistochemistry while the neuroendocrine tumor of the appendix normal expression of MMR proteins. This discrepancy showed that the two tumors arose through different molecular pathways and that the NET is not related to the germline deletion in the MLH1 gene.
In conclusion, the frequency of de novo PVs in LS remains to be established; however, the application of universal screening for loss of MMR function in all newly diagnosed patients with CRC and endometrial cancer would improve their detection. De novo PV identification is crucial to allow proper genetic counseling and to avoid unnecessary screening in relatives. Further studies are needed to explore the possible mechanisms involved in the occurrence of de novo PVs in LS.
Supplemental Material
sj-pdf-1-tmj-10.1177_03008916231197113 – Supplemental material for De novo germline pathogenic variant in Lynch Syndrome: A rare event or the tip of the iceberg?
Supplemental material, sj-pdf-1-tmj-10.1177_03008916231197113 for De novo germline pathogenic variant in Lynch Syndrome: A rare event or the tip of the iceberg? by Clorinda Brignola, Sara Volorio, Giovanna De Vecchi, Daniela Zaffaroni, Valentina Dall’Olio, Frederique Mariette, Domenico Sardella, Fabio Capra, Stefano Signoroni, Emanuele Rausa, Marco Vitellaro, Valeria Pensotti and Maria Teresa Ricci in Tumori Journal
Footnotes
Acknowledgements
The authors wish to acknowledge and express sincere gratitude to Mariangela Di Ceglie, Ornella Galuppo and Antonio Zaccara from the Unit of Hereditary Digestive Tract Tumors for prospectively maintaining the patients’ database.
Author contributions
Clorinda Brignola, Giovanna De Vecchi, Valeria Pensotti, Maria Teresa Ricci designed and coordinated the study, recruited the family and drafted the manuscript. Sara Volorio, Valentina Dall’Olio, Frederique Mariette, Domenico Sardella, Fabio Capra processed blood samples, conducted the experiments and performed bioinformatic analysis. Daniela Zaffaroni, Stefano Signoroni, Emanuele Rausa, Marco Vitellaro collected the clinical data and modified the manuscript. All authors critically reviewed the manuscript, approved the final version, and are accountable for all aspects of the work.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.