
Abstract
Lynch syndrome is a genetic condition predisposing to cancer, particularly colorectal cancer and endometrial cancer, due to germline mutations in MisMatch Repair genes. More rarely, Lynch syndrome is the result of a constitutional MLH1 promoter methylation. This review summarizes the current knowledge about the role of this epigenetic mechanism in the Lynch syndrome. Universal Tumor Screening, performed on tumoral specimens to identify features suggestive of Lynch syndrome, shows the same features both in the case of sporadic cancers and Lynch syndrome-cancers due to a constitutional MLH1 methylation: microsatellite instability, deficiency of MisMatch Repair proteins, and methylation of MLH1 gene. Over the last few years, identifying methylation of MLH1 promoter on tumors was used to discern sporadic tumors from Lynch syndrome tumors: the methylation of the MLH1 promoter was usually explained as a somatic event and this could lead to a missed diagnosis of some Lynch syndrome cases. Therefore, establishing criteria to decide when to test patients for constitutional MLH1 methylation is urgent. In the case of microsatellite instability/deficiency of MisMatch Repair tumors with MLH1 methylation, a germline genetic test could be requested for all colorectal cancer patients aged 55 years or younger and all endometrial cancer patients younger than 50 years old, independently from family history. The prevalence of germline MLH1 epimutations is not precisely known and possibly underestimated. The associated cancer risk could be similar to that due to a MLH1 sequence variant. MLH1 epimutations could be secondary to other genetic defects and follow an autosomal dominant inheritance. On the contrary, primary epimutations are often “de novo” events, and their transmission does not follow Mendelian rules.
Keywords
Introduction
Lynch syndrome (LS) is a well-known condition predisposing to cancer, particularly colorectal cancer (CRC) and endometrial cancer (EC), with an autosomal dominant inheritance pattern. Heterozygous germline pathogenic (or likely pathogenic) sequence variants or large rearrangements in one of the DNA MisMatch Repair (MMR) genes (MLH1, MSH2, MSH6, and PMS2) are the most frequent causes of LS (about 90% of LS are attributable to this mechanism). Two epigenetic mechanisms are a less frequent cause of LS: germline deletions of the EPCAM gene (responsible for the silencing of MSH2 gene) and constitutional methylation of MLH1 promoter. 1 Constitutional methylation of the MLH1 promoter in one of the two alleles of the gene causes the silencing of that allele by a mechanism of transcriptional downregulation. 2 Monoallelic silencing of MLH1 expression due to constitutional methylation of MLH1 promoter is a cancer-predisposing element because it is the first hit in the tumorigenesis mechanism. 3
Identifying patients with LS caused by constitutional methylation of the MLH1 promoter is a current issue. Nowadays, somatic analyses included in the so-named Universal Tumor Screening (UTS) are carried out in all CRCs to identify patients with LS. MLH1 promoter methylation detected on tumor samples reduces the likelihood of a genetic predisposition, being more frequently referable to somatic events. Accordingly, in the case of tumor samples with MLH1 methylation, germline tests are not performed unless a remarkable family history or an early age of onset is present. 4 However, this approach does not completely exclude the possibility of missing some LS patients with constitutional MLH1 methylation. So, establishing new criteria to suspect LS in the case of tumor samples with MLH1 promoter methylation is an urgent question. 5
This review aims to summarize the current knowledge about constitutional MLH1 promoter methylation while seeking answers to some questions: when to test for constitutional epimutations of MLH1? What is the prevalence of germline methylation of the MLH1 gene? What is the associated cancer risk? How does the inheritance of MLH1 epimutations work?
Lynch syndrome diagnosis and MLH1 promoter methylation
In the past, the only tools for identifying LS patients were clinical criteria, such as Amsterdam criteria and Bethesda revised guidelines (rBG) (Table 1).6 -9 The introduction of the rBG increased the sensibility compared to the Amsterdam criteria while reducing the specificity. However, the sole use of clinical criteria implicates a non-negligible risk of missing about 28% of LS patients.4,6
Clinical Criteria.

CRC, colorectal cancer; EC, endometrial cancer; LS, Lynch syndrome; MSI, microsatellite instability.
Currently, to better identify LS patients, UTS should be performed in all new cases of CRC (and also in all ECs) regardless of family and personal cancer history.4,10,11 UTS involves the evaluation of the MMR system by the determination of microsatellite instability (MSI) and/or the execution of immunohistochemical assays to assess the expression of MMR proteins (IHC-MMR). 11 Microsatellites are in tandem repeated sequences of di/tetra-nucleotides, susceptible to length variations because of pairing mistakes during replication. The function of MMR proteins is to avoid the accumulation of these mistakes. 12 Typically, LS-related cancers show MSI and/or loss of immunohistochemical expression of MMR proteins as a consequence of their MMR-deficiency (d-MMR) 11 ; d-MMR is caused by biallelic pathogenic variants in the MMR genes (according to Knudson’s two-hit model, the first variant is germline, while the second is a somatic acquired defect).3,4 The determination of MSI and immunohistochemistry in CRCs is usually concordant. However, the two tests combined increase the sensibility of the UTS. 11 Of note, IHC-MMR points to the most likely defective genes. Differently, MSI only highlights the presence of a possible genetic defect in MMR genes without pointing to the specific involved gene and requiring a multigene panel test including all five LS-related genes.
About 13-15% of screened CRCs present d-MMR, 11 and more than 70% of CRCs with d-MMR are sporadic tumors 4 due to somatic methylation (and consequent inactivation) of the MLH1 gene. 11 Less frequently, the methylation of the MLH1 promoter represents the second hit, which leads to the development of cancer in a carrier of a germline pathogenic or likely pathogenic variant of the MLH1 gene. Even more rarely, it is the first hit in a carrier of an MLH1 epimutation (i.e., constitutional methylation of the MLH1 promoter).
The UTS algorithm (Figure 1) aims to facilitate the identification of LS-related cancers. 4 When the UTS shows a loss of PMS2, MSH2, or MSH6 expression, the germline test is always recommended according to IHC-MMR results. UTS algorithm recommends an intermediate step in the case of MLH1 loss of expression (both in the rare case of an isolated MLH1 loss of expression and in the more common case of the detection of loss of MLH1 expression together with loss of PMS2 expression): the search for the somatic BRAF V600E variant and the analysis of MLH1 promoter methylation. When the somatic BRAF V600E variant and the MLH1 promoter methylation are absent, a germline analysis of MLH1 and PMS2 genes is recommended. Otherwise, if one of these tests is positive, LS is highly unlikely unless a remarkable personal and/or family history is present (Figure 1). According to the last NCCN guidelines, 13 in cases with young age of onset (<50 years) or with a significant personal and/or family history, a germline test of all MMR genes (sequence variants and large rearrangements) or for constitutional MLH1 epimutation is recommended. However, although age of onset and family history are the most promising criteria, there are still many open questions regarding how to use them. 5
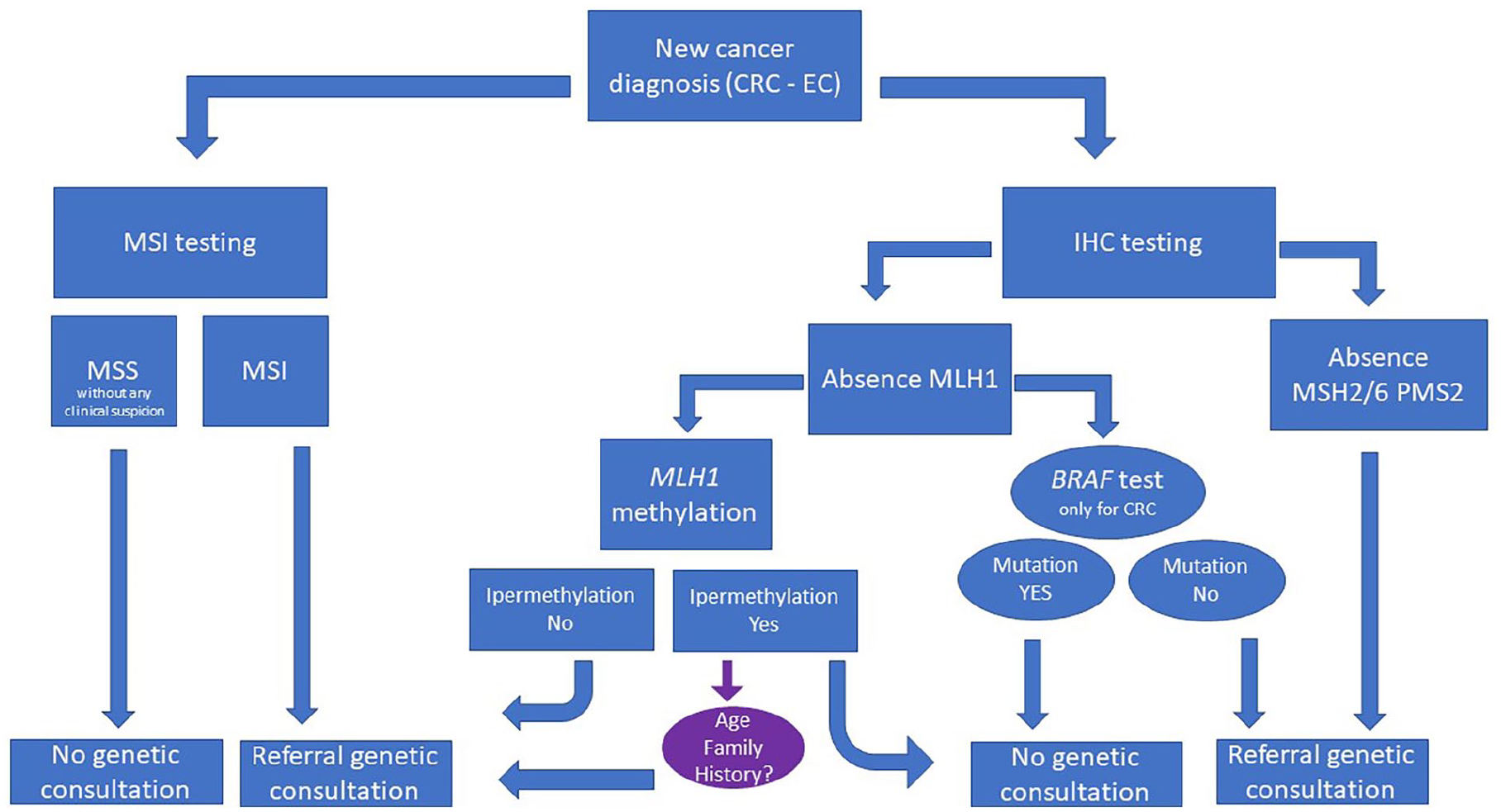
Universal Tumor Screening algorithm.
In addition to the risk of developing CRC, women with LS have a high lifetime risk of developing EC. Notably, the UTS is now recommended for ECs, as well as for CRC. About 20-30% of ECs show dMMR/MSI, most of which are sporadic cancers. If IHC-MMR indicates loss of MLH1 and/or PMS2 protein expression, the analysis of MLH1 methylation on the tumor sample is requested. Contrary to CRCs, in ECs the search for somatic V600E BRAF mutation is not recommended. 11 The methylation of MLH1 promoter is not sufficient to rule out LS. Therefore, especially in patients with a suggestive personal and/or family history, searching for germline mutations in MMR genes and constitutional MLH1 epimutations could be advisable. 14
Somatic MLH1 promoter methylation as second hit in patients with LS
The presence of MLH1 methylation in the tumor sample does not allow us to rule out with absolute certainty the possibility of LS. Cancers exhibiting MLH1 methylation can hide an MLH1 constitutional epimutation that could be the first hit in tumorigenesis. Furthermore, in a small, but not negligible, number of cases somatic MLH1 methylation could be the acquired second hit in the process of tumorigenesis in a patient who carries a sequence variant causative for LS: somatic MLH1 methylation could affect the wild-type MLH1 allele and work as the second hit in the tumorigenesis of carriers of MLH1 germline sequence variant.15,16
About 15% of CRCs developed by LS patients who are carriers of a sequence variant exhibit MLH1 methylation.15,17,18 Different levels of MLH1 methylation, even in patients with the same sequence variant, can be considered evidence that MLH1 methylation is not due to the variant itself but is an acquired defect that occurs during tumorigenesis. 15 Somatic MLH1 methylation has been described as the second hit of tumorigenesis in LS patients even in cases of ECs, maybe even more frequently than in CRCs, but larger studies are needed to confirm this hypothesis. 15 Methylation of MLH1 promoter is described in the tumor samples not only of carriers of constitutional MLH1 sequence variant but also in patients with LS due to sequence variants in one of the other MMR genes. A recent work by Helderman and collaborators 16 reports the presence of MLH1 methylation in cancers of carriers of a sequence variant in MLH1, MSH2, MSH6, and PMS2 genes. At the molecular level, in carriers of MSH2, MSH6, or PMS2 germline variants, the route of tumorigenesis could be difficult to understand precisely, and further studies are needed; one hypothesis is that a second (acquired) hit in the MSH2, MSH6, or PMS2 gene could be followed by biallelic somatic methylation of MLH1. 16
To summarize, when LS-related cancers exhibit MLH1 hypermethylation, it could be somatic or constitutional. In particular, three different possibilities explain the presence of MLH1 hypermethylation in LS-related cancers:
- MLH1 hypermethylation identified in cancer could be the acquired second hit in the tumorigenesis in a patient with LS due to a germline sequence variant (first hit)
- somatic MLH1 hypermethylation could be the acquired second hit in a patient with LS due to a constitutional MLH1 hypermethylation (first hit)
- MLH1 hypermethylation identified in cancer could be a manifestation of a condition of LS due to a constitutional MLH1 hypermethylation; in this case a somatic sequence variant could be the second hit.
Thus, in suspected cases, testing for constitutional MLH1 epimutations and sequence variants in MMR genes is highly recommended.
Prevalence of constitutional MLH1 promoter methylation
The detection rate of both primary and secondary MLH1 epimutations significantly increases in patients selected according to their personal and/or family history. In a cohort of patients who fulfill the rBG, the detection rate for constitutional epimutations is up to 15,6%. 19 On average, the prevalence of constitutional methylation of the MLH1 promoter is about 10% in cases selected not only according to the tumoral features but also for fulfilling the rBG or for having suspicious clinical features. On the contrary, in cohorts of patients selected only according to the tumoral features, the prevalence is low, although not yet precisely estimated.2,19 -21
Cancer risk associated with constitutional MLH1 promoter methylation
An open question is cancer risk assessment for patients with constitutional MLH1 methylation. When LS is due to a sequence variant, cancer risk varies according to the involved MMR gene. Although there is still a debate about the effective involvement of some MMR genes in specific cancer risks (e.g., PMS2 and ECs), risk estimations are available for each MMR gene in the literature. 22 On the contrary, the first patients with constitutional MLH1 epimutations described in scientific literature came from high-risk families or presented suspicious personal features (such as the fulfillment of clinical criteria), leading to possible bias in estimating their risk of cancer. Furthermore, to the best of our knowledge, only numerically small cohorts of unselected patients with constitutional MLH1 epimutation have been described so far.
It could be reasonable to assume that constitutional MLH1 epimutations confer cancer risks similar to carriers of a sequence variant in the MLH1 gene. Patients with a germline MLH1 epimutation have a high personal risk of developing multiple metachronous or synchronous Lynch-related cancers at a young age, regardless of family history.21,23. As expected, the main risks are for early onset CRC and EC. 2 The mean age of onset for CRC is about 37-40 years with a very broad range (from 17 to 69 years). 24 Moreover, other Lynch-related cancers are described, such as urothelial, pancreatic, gastric and even breast cancer. 24
However, further studies on larger cohorts are needed to estimate the associated cancer risks. While waiting for detailed knowledge about the risk assessment, patients with constitutional methylation of MLH1 should be managed as patients with a sequence variant in the MLH1 gene. 2
Inheritance of MLH1 epimutation
Genetic counseling for issues related to transmission and testing of at-risk relatives is challenging for MLH1 epimutations. Indeed, epimutations can be distinct into “secondary” or “primary” epimutations.
A “secondary” epimutation is generally due to a sequence variant causing the epimutation. The epimutation and the causative sequence variant cosegregate, and the transmission follows autosomal dominant inheritance; in these cases, an informative family history is usually present. 25 What is transmitted from one generation to the next is the sequence variant responsible for the epimutation. Usually, the sequence variant is cis-acting and localized in a non-coding region nearby the gene sequence. 24 Generally, the first alteration identified in the proband is the constitutional methylation, then it is possible to search for the non-coding sequence variant. A sequence variant is not always identified, even in families in which it seems that the epimutation defect could follow an autosomal dominant manner of inheritance. However, it could not be excluded that the lack of an evident sequence variant depends on the limits of our knowledge: the presence of a distant cis-acting, or even trans-acting, sequence alteration cannot be ruled out. 26
An epimutation is “primary” when there is no evidence of a sequence variant responsible for the epimutation itself, and the inheritability pattern seems not to follow an autosomal inheritance pattern. These constitutional epimutations tend to arise as a de novo event, so the family history is usually not informative. The transmission of a “pure” (“primary”) epigenetic defect to the next generation does not follow Mendelian rules.24,27 During gametogenesis and the first phases of the embryonal development, there is an epigenetic reprogramming in which the correction of epimutation defects is possible (reversibility of the epimutation): a methylated MLH1 allele can be transmitted to the offspring in an unmethylated form. 27 The erasure and reprogramming of the epigenetic marks in the passage from one generation to the next are not infallible: epimutations can resist the reprogramming, and so they can be transmitted to the next generation following a non-Mendelian pattern of inheritance. In these cases, the risk of transmission is “low” (much less than the 50% associated with dominant inheritance), although it exists and is not precisely quantifiable. 27 Epimutations could be more frequently maintained during oogenesis than during spermatogenesis. However, further studies are needed to verify this hypothesis. Indeed, paternal transmission could not be excluded since cases with low levels of MLH1 methylation in DNA from spermatozoa have been described. 27
In the case of a “primary” epimutation, all the first-grade relatives (and the other relatives following the cascade system test) should be tested to verify the germline state of MLH1 promoter methylation. Similarly, in the case of a “secondary” epimutation, a genetic test should be offered to relatives to verify the presence of the constitutional MLH1 epimutation and, when known, the germline sequence variant responsible for the MLH1 methylation. Usually, tests to search for a MLH1 epigenetic defect and/or a MMR gene sequence variant in relatives of LS patients are recommended for individuals older than 18 years.
Mosaic MLH1 methylation
“Mosaicism” occurs when there are two different cellular lines in the same organism. The MLH1 epimutations could be present in the form of mosaicism. 28 The personal oncological risk of patients with mosaicism depends on the percentage of mosaicism (how many cells of the whole organism present MLH1 epimutation) and on the somatic distribution (the risk increases if the MLH1 epimutation is present in Lynch-related organs). These two aspects are both challenging to estimate. Mosaicism is a de novo post-zygotic event. Thus, in these cases, there is no need for a segregation analysis in the parents of the proband or genetic analysis in siblings. Otherwise, the MLH1 epimutation should be searched for in the offspring of the proband because the possibility of transmission exists in this case. However, recurrence risk in offspring is difficult to quantify because it depends on the presence and the percentage of gonadic mosaicism in the proband. 28
When to test constitutional MLH1 promoter methylation
In the presence of a tumor sample with the MLH1 promoter methylation, family history could be a helpful criterion to identify carriers of a pathogenic or likely pathogenic variant of the MLH1 gene or secondary MLH1 epimutations. On the contrary, this criterion may be useless to detect primary MLH1 epimutation for two reasons: 1) constitutional methylation of MLH1 often arises as a “de novo” event, so the family is not informative; 2) transmission of constitutional MLH1 methylation does not always follow the Mendelian rules.
Therefore, the age of onset is the most promising criterion for detecting the primary MLH1 epimutations. However, there is a need to establish the best cut-off for age of onset to require a germline MLH1 promoter analysis. According to a recent paper by Hitchins et al., 5 in case of a CRC with MSI/dMMR and methylation of the MLH1 promoter, the germline MLH1 promoter analysis should be required in all patients aged 55 years or younger, independently from family or personal history. They tested two cohorts of 95 and 281 CRC patients, respectively; patients were retrospectively selected only on the basis of the tumoral features: MSI/dMMR and methylation of the MLH1 promoter. They found and described eight cases (2%) with constitutional methylation of the MLH1 promoter. As expected, they found that the prevalence of germline MLH1 methylation is higher at younger ages, particularly for patients ⩽55 years: in this group it was detected in about 33% (7/21) of cases. They concluded that constitutional MLH1 methylation should be investigated in all patients ⩽ 55 years and with CRC dMMR/MSI with MLH1 promoter methylation. 5
Regarding ECs, in a recent study, Hitchins and colleagues studied the prevalence of constitutional MLH1 methylation in a cohort of ECs selected according to their tumoral features (dMMR and somatic MLH1 methylation). The prevalence of constitutional MLH1 epimutation was about 17% and 2% in the cohort with ECs diagnosed <50 and <60 years, respectively, contrary to the 0-1% in unselected ECs. Thus, they recommended testing patients <50 years with EC showing a somatic MLH1 methylation and all patients with multiple (synchronous or metachronous) LS-related tumors with MLH1 methylation, regardless of age or family history. 29
Conclusion
Even if the methylation of the MLH1 promoter detected on tumor samples is a typical feature of sporadic cancers, nowadays, it is well-known that in a minority of cases it could hide the presence of LS. Since the existence of methylation of the MLH1 promoter and the diagnosis of LS are not mutually exclusive, MLH1 methylation could not be used to rule out the possibility of LS. Indeed, MLH1 methylation could be the first hit of tumorigenesis in a patient with germline MLH1 epimutation, or it could be the second hit of tumorigenesis in a patient with LS due to a germline sequence variant in the MMR genes. The diagnosis of LS in a patient with cancer showing dMMR and MLH1 methylation is rare. However, it is possible and has relevant consequences in the clinical management of the proband and the family.
According to recent works by Hitchins et al., in the case of MSI/dMMR tumors with MLH1 methylation, a germline genetic test could be performed for all CRC patients aged 55 years or younger and for all EC patients younger than 50 years, independently from family history.5,29
There are still many open questions and challenges about constitutional MLH1 epimutations and their management; the prevalence of MLH1 epimutation is low and not precisely esteemed, the oncological risk of carriers of MLH1 constitutional epimutation has not been exactly quantified, transmission does not strictly follow Mendelian rules and the risk of transmission to the offspring could be difficult to estimate.
Together with the clinical criteria (suggestive personal and/or family history), the UTS greatly supports reaching a more comprehensive LS diagnosis. To further improve this achievement, it is now important to define criteria to test for constitutional MLH1 epimutation.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.