
Abstract
Objective
Primary bilateral macronodular adrenal hyperplasia (PBMAH) is a rare cause of Cushing syndrome. Recent evidence, particularly the 2023 European Society of Endocrinology guidelines, clinically classifies cortisol excess into overt Cushing syndrome and mild autonomous cortisol secretion. Germline mutations in the Armadillo repeat-containing 5 (ARMC5) gene have been identified in 20%–55% of patients with PBMAH. This study aimed to describe the clinical, radiological, and biochemical characteristics of PBMAH; classify cortisol secretion according to updated guidelines; and evaluate surgical and conservative treatment outcomes.
Methods
This retrospective cohort study included 58 patients with bilateral adrenal macronodules who underwent ARMC5 genetic testing at a tertiary center between 2023 and 2025. Clinical, imaging, laboratory, and genetic data were collected. Mild autonomous cortisol secretion was defined according to the 2023 European guidelines as a post–dexamethasone suppression test cortisol level >1.8 μg/dL in the absence of overt Cushing syndrome features. Postoperative and follow-up data, including hormonal assessments and remission criteria, were recorded.
Results
A total of 58 patients were included; 14 (23.7%) were male, and the mean age was 57.7 years (39–73). Four patients (6.9%) with overt Cushing syndrome carried germline ARMC5 mutations, three of whom belonged to the same family, supporting an autosomal dominant pattern of inheritance. Among them, 34 patients with mild autonomous cortisol secretion and 20 patients with nonfunctional PBMAH were managed conservatively. Unilateral laparoscopic adrenalectomy was performed in five patients, all of whom achieved biochemical and clinical remission during follow-up (median, 14 months). No postoperative adrenal insufficiency or persistent hypercortisolism was observed.
Conclusion
PBMAH demonstrates a broad clinical spectrum, ranging from nonfunctional disease to overt Cushing syndrome. The updated classification of cortisol secretion (mild autonomous cortisol secretion vs. overt Cushing syndrome) improves clinical stratification and supports treatment decision-making. ARMC5 genetic analysis contributes to diagnostic confirmation, facilitates cascade family screening, and enables identification of asymptomatic carriers. Unilateral adrenalectomy is effective in patients with overt Cushing syndrome, whereas surveillance is appropriate for those with mild autonomous cortisol secretion or nonfunctional PBMAH.
Keywords
Introduction
Primary bilateral macronodular adrenal hyperplasia (PBMAH) is characterized by bilateral benign adrenal macronodules larger than 1 cm, with variable degrees of cortisol excess. 1 PBMAH is a rare cause of Cushing syndrome (CS), accounting for <2% of all cases. 2 Most PBMAH cases are detected incidentally during abdominal computed tomography (CT) or magnetic resonance imaging (MRI) performed for unrelated conditions.
In 2022, the World Health Organization (WHO) introduced a revised classification of adrenal cortical nodular disease. 3 Adrenal nodular disorders are currently categorized as sporadic nodular adrenal disease, bilateral micronodular adrenal disease, and bilateral macronodular adrenal disease. According to the 2023 European Society of Endocrinology guidelines, adrenal hyperplasia is classified into two main forms: the diffuse type, in which prominent nodules are absent, and the macronodular type, in which multiple nodules are typically present in both adrenal glands (usually with a Hounsfield unit (HU) <10 on noncontrast imaging) and the remaining adrenal tissue is generally thickened. Based on biochemical findings and the presence of clinical features, cortisol excess is stratified into mild autonomous cortisol secretion (MACS) and overt CS. 4 These updated definitions improve diagnostic standardization and align disease classification with contemporary clinical practice.
Historically considered a sporadic disorder, PBMAH is now recognized as frequently having a genetic basis. In 2013, germline mutations in the Armadillo repeat-containing 5 (ARMC5) gene were identified as a major contributor to PBMAH with an autosomal dominant pattern of inheritance. 5 Subsequent studies have demonstrated that ARMC5 alterations function as tumor suppressor defects and are detected in 20%–55% of affected patients; mutation carriers exhibit earlier disease onset, larger adrenal nodules, more pronounced cortisol secretion, and more severe disease.6,7 Familial clustering, variable expressivity, and incomplete penetrance further support autosomal dominant inheritance and suggest the influence of genetic or environmental modifiers. 8
At the molecular level, ARMC5 encodes a scaffold protein that participates in ubiquitin ligase complexes, facilitating ubiquitination and proteasomal degradation of substrates such as DNA-dependent RNA polymerase II (RPB1). Loss-of-function mutations reduce RPB1 ubiquitination, resulting in its accumulation, dysregulated gene expression, and nodular adrenal growth. 9 ARMC5 deficiency also disrupts apoptosis, enhances adrenal cortical cell proliferation, and alters steroidogenic pathways, thereby contributing to cortisol excess.5,8
Clinically, ARMC5-related PBMAH exhibits a broad spectrum ranging from MACS to overt CS. Genetic testing is recommended in patients with bilateral adrenal macronodules or those with a family history of adrenal disease, enabling early identification of mutation carriers and guiding surveillance and therapeutic decision-making. Although steroidogenic capacity per cell is reduced, the overall increase in adrenal mass results in net cortisol overproduction. This explains why PBMAH often remains asymptomatic until later adulthood, typically between 40 and 70 years.10,11
The clinical presentation ranges from asymptomatic disease to overt CS, depending on the degree of cortisol excess. PBMAH may be rarely associated with autonomous aldosterone or androgen secretion. Although definitive diagnosis is established by histopathological evaluation following adrenalectomy, diagnosis in nonoperated patients relies on radiological imaging and biochemical assessment. 12
Metabolic comorbidities, including hypertension, diabetes mellitus, and obesity, are common, whereas classic stigmata of CS, including rapid weight gain, supraclavicular and dorsocervical fat accumulation, and violaceous striae, are less frequent. Determining which patients require intervention and selecting the optimal management strategy remain major clinical challenges. 11 In the absence of overt clinical features of CS, patients with dexamethasone suppression test (DST) value of >1.8 μg/dL are considered to have MACS. Medical management of associated comorbidities (hypertension, diabetes mellitus, dyslipidemia, and osteoporosis) is recommended for asymptomatic and subclinical patients. However, patients with severe or progressive comorbidities are candidates for surgical intervention. In such cases, unilateral adrenalectomy of the larger gland is recommended. When remission is not achieved, partial or total resection of the contralateral adrenal gland may be considered. Despite achieving hormonal control, bilateral adrenalectomy results in lifelong adrenal insufficiency and is associated with increased perioperative mortality (6%–8%). 13 In contrast, unilateral adrenalectomy achieves remission in approximately 60% of cases, with substantially lower morbidity. 14
In this study, we aimed to characterize the clinical features, updated cortisol-secretion classification, radiological findings, genetic results, and treatment outcomes of patients with PBMAH managed at our tertiary endocrine center.
Materials and methods
This retrospective cohort study included 58 patients with bilateral adrenal macronodules who underwent ARMC5 genetic testing at a tertiary center between 2023 and 2025. Clinical, radiological, and biochemical data along with information on associated comorbidities were obtained from electronic medical records. The study was conducted in accordance with the Declaration of Helsinki (2024 update) and was approved by our institutional ethics committee (Approval Number: 150/2025). All patient data were fully deidentified. The reporting of this study conforms to the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) guidelines. 15
The diagnosis of PBMAH was established according to the 2023 European Society of Endocrinology guidelines, based on radiological identification of bilateral adrenal macronodules >1 cm and thickening of the remaining adrenal tissue on CT and/or MRI. Clinical features of overt CS, adrenal nodule morphology, and biochemical findings, including the 1-mg overnight DST (μg/dL), 24-h urinary free cortisol (UFC; μg/24 h), serum cortisol (μg/dL), adrenocorticotropic hormone (ACTH; pg/mL), and aldosterone (ng/dL), were recorded.
Cortisol secretion status was classified according to the 2023 European Society of Endocrinology guideline as follows:
Overt CS. Presence of characteristic clinical features and an abnormal DST result; MACS. Post–1-mg DST cortisol concentration >1.8 μg/dL in the absence of overt CS features; Nonfunctional. Post–1-mg DST cortisol concentration ≤1.8 μg/dL.
Genetic testing was performed using a targeted next-generation sequencing (NGS) panel on peripheral blood samples. The panel included ARMC5, the gene most frequently implicated in PBMAH. KDM1A was not included in the panel; therefore, patients could not be evaluated for KDM1A-associated disease or food-dependent CS. This limitation is addressed in the Discussion section. Family history was systematically assessed, and cascade genetic testing of first-degree relatives was recommended for patients with pathogenic ARMC5 variants; three relatives underwent testing.
Patients were managed either surgically or conservatively based on clinical, radiological, and biochemical findings. Indications for surgery included overt CS, clinically significant comorbidities (e.g. diabetes mellitus and hypertension) in patients with MACS, and patient preference. Radiological and biochemical evaluations were performed during follow-up.
Postoperative follow-up included assessment of serum cortisol, ACTH, DST, and UFC at the first postoperative month. When cortisol levels were found within the normal range, annual follow-up was continued. However, when cortisol levels were found to be below or above the normal range, appropriate medical treatment was initiated, and follow-up evaluations were conducted at the third, sixth, and ninth months after surgery. Clinical remission was defined as resolution of CS features and normalization of biochemical parameters.
Results
A total of 58 patients with bilateral adrenal macronodules were included. Of them, 14 (23.7%) were male, and the mean age was 57.7 years (range, 39–73). All patients met the radiological criteria for PBMAH. PBMAH was detected in 38 patients (65.5%) via MRI and in 48 patients (82.7%) via CT. According to imaging findings, nodule sizes ranged from 10 to 60 mm (Figure 1).
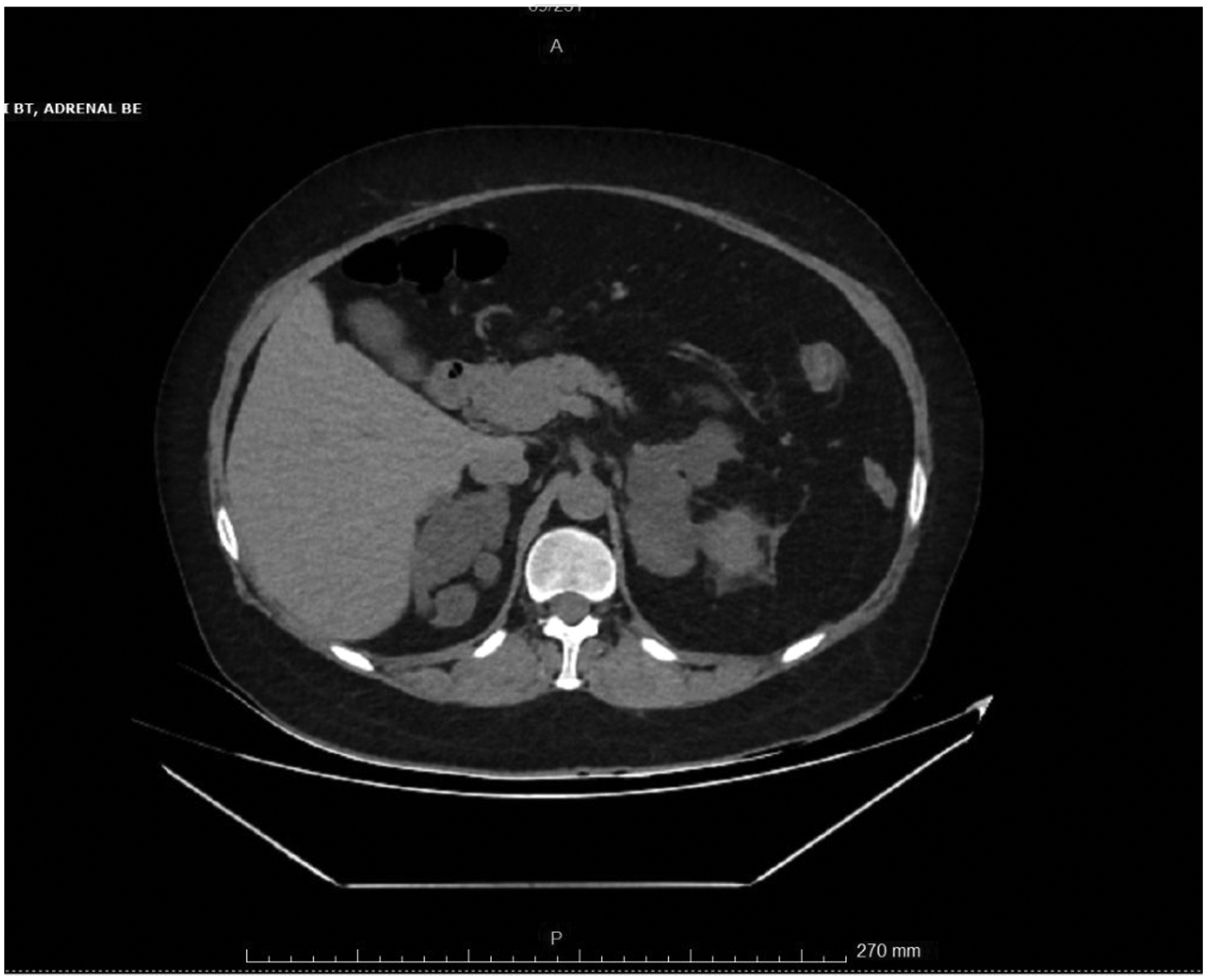
Bilateral macronodular adrenal hyperplasia on CT imaging: diffuse thickening and contour lobulation on bilateral adrenal glands. The lateral crus thickness was 28 mm and the medial crus thickness was 19 mm in the right adrenal gland. The lateral crus thickness was 45 mm and the medial crus thickness was 36 mm in the left adrenal gland. CT: computed tomography.
Clinically, 4 patients (6.8%) had overt CS. Among the remaining 54 patients, 34 (58.7%) were classified as having MACS and 20 (34.5%) as nonfunctional based on a DST cutoff of >1.8 μg/dL. DST values were ≥1.8 μg/dL in 38 patients (65.5%), whereas the remaining 20 patients had values below this threshold and were followed conservatively without treatment (Table 1).
Demographic, imaging, and hormonal characteristics of the study population.
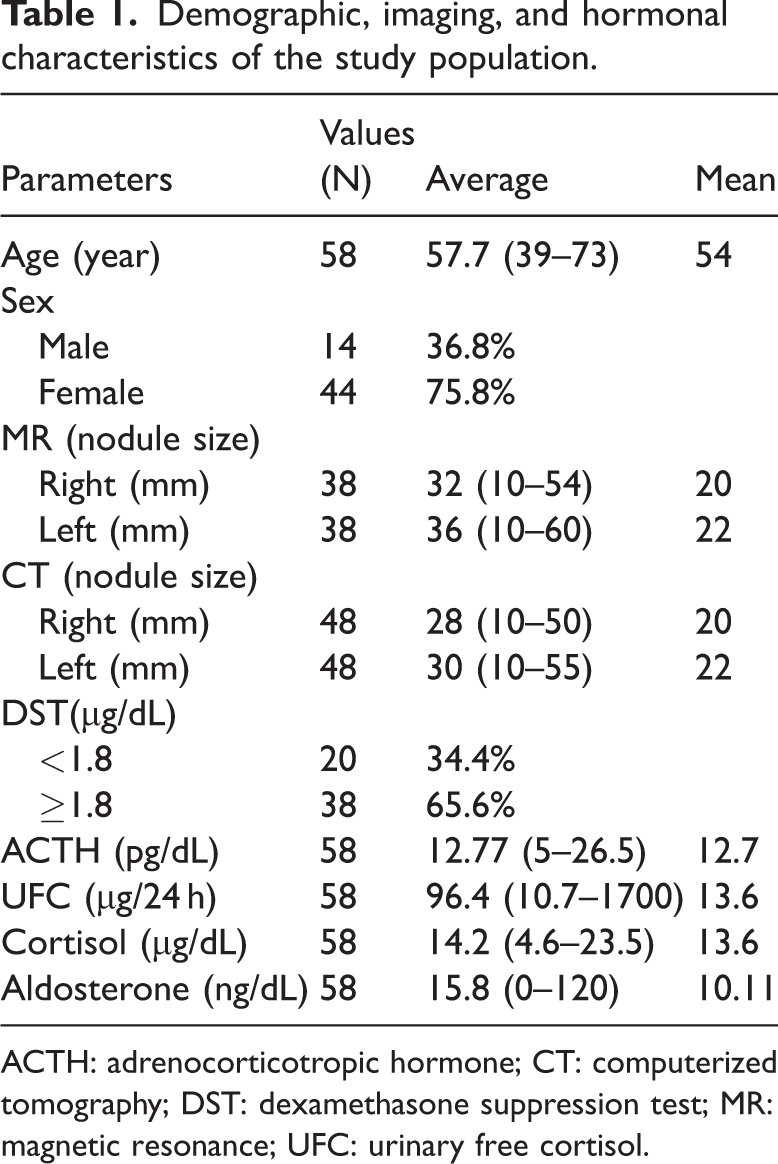
ACTH: adrenocorticotropic hormone; CT: computerized tomography; DST: dexamethasone suppression test; MR: magnetic resonance; UFC: urinary free cortisol.
Mean biochemical values were as follows: ACTH, 12.77 pg/mL (range, 5–26.5); UFC, 96.4 μg/24 h (range, 10.7–1034); and serum cortisol, 14.2 μg/dL (range, 4.6–23.5). The mean aldosterone level was 15.8 ng/dL (range, 0–120.01). All demographic, biochemical, and radiological characteristics are summarized in Table 1.
Pathogenic ARMC5 variants were identified in four patients (6.8%). All mutation carriers had overt CS, and three belonged to the same family, supporting an autosomal dominant pattern of inheritance. Three of these patients were female. Their detailed clinical and radiological findings are presented in Table 2. Although statistical analysis was limited by the relatively small sample size, ARMC5-positive patients demonstrated higher cortisol levels and larger nodules on average, consistent with previous reports.
Laboratory findings of ARMC5-positive patients with overt CS.

ACTH: adrenocorticotropic hormone; ARMC5: Armadillo repeat-containing 5; CS: Cushing syndrome; UFC: urinary free cortisol.
Comorbidities were present in 55 patients (93.2%), most commonly hypertension (n = 29, 50.0%), diabetes mellitus (n = 18, 31.0%), and hyperlipidemia (n = 15, 25.8%). Metabolic comorbidities were more prevalent in patients with overt CS and MACS than in those with nonfunctional disease. In 26 of 38 patients (68.4%) with DST levels >1.8 μg/dL, hypertension, diabetes mellitus, and hyperlipidemia were diagnosed, either separately or in combination. Among the 20 patients with nonfunctional disease, 10 (50.0%) had metabolic comorbidities.
Laparoscopic unilateral adrenalectomy, a low-morbidity surgical option increasingly recommended in recent years, was performed in four patients with overt CS and in one patient with MACS who had clinically significant comorbidities. Pathological examination confirmed macronodular adrenal cortical hyperplasia in all cases. The remaining 33 patients with MACS and 20 patients with nonfunctional disease were managed conservatively.
The mean follow-up duration was 14 months (range, 6–30 months). None of the surgically treated patients developed recurrence during follow-up; all achieved biochemical normalization and complete resolution of clinical CS features. Adrenal insufficiency was not observed in any patient who underwent surgery (Table 3). Figures 2 and 3 present preoperative and postoperative photographs of one patient with overt CS who experienced >20 kg weight loss following surgery.
Postoperative laboratory findings of the patients undergoing unilateral adrenalectomy.

ARMC5: Armadillo repeat-containing 5; ACTH: adrenocorticotropic hormone; ald: aldosterone; DST: dexamethasone suppression test; F: female; S: sex; M: male; Neg: negative; Posi: positive; Postop: postoperative; Preop: preoperative; UFC: urinary free cortisol.
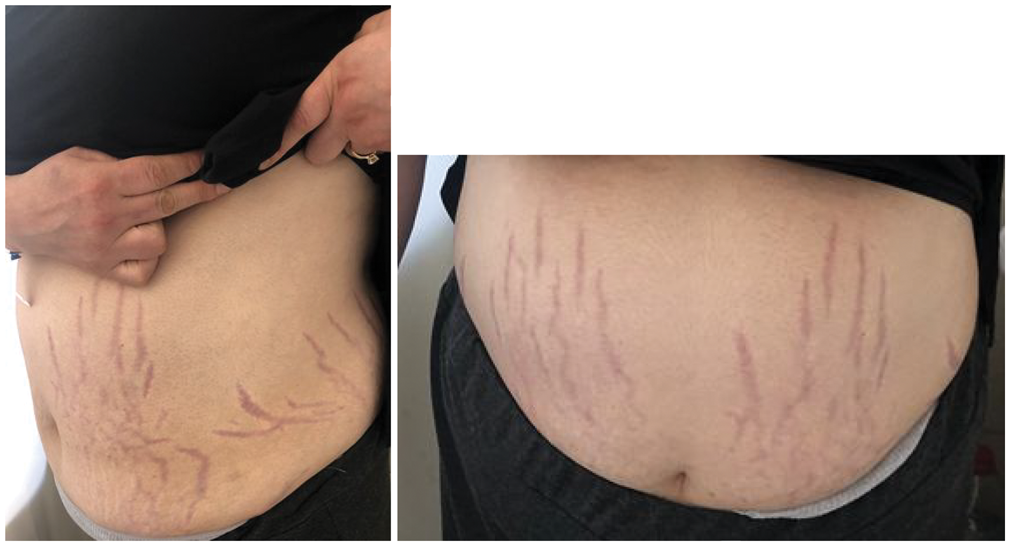
Stria of the abdominal skin in a patient with Cushing syndrome (preoperative).

Disappearance of stria after unilateral adrenalectomy.
Discussion
This single-center cohort study provides a comprehensive evaluation of the clinical, biochemical, radiological, and genetic characteristics of 58 patients with PBMAH diagnosed at a tertiary endocrine referral center. PBMAH is a rare but increasingly recognized cause of ACTH-independent CS. Following the identification of ARMC5 mutations in 2013, the hereditary basis of PBMAH has become more clearly established.
In our cohort, ARMC5 mutations were detected in 6.8% of patients and in 10.5% after exclusion of the nonfunctional PBMAH group. These prevalence rates are lower than those reported in previous studies (25%–55%)5,8,16 and may be explained by differences in patient selection, the inclusion of a large proportion of MACS and nonfunctional cases, the use of a limited sequencing panel, and possible ethnic or geographic variation. Notably, three of the four ARMC5-positive patients belonged to the same family, underscoring the importance of cascade genetic testing and family counseling. All identified families were offered genetic counseling to facilitate screening of at-risk relatives and to provide education regarding inheritance patterns and potential clinical manifestations.
PBMAH exhibits marked clinical heterogeneity. Sporadic PBMAH is reported to occur 2–3 times more frequently in females than in males, whereas familial PBMAH affects both sexes equally.17,18 In our study, three of the four ARMC5-positive patients were female, consistent with the predominance observed in sporadic cases; however, familial clustering was also observed. The mean age of the cohort was 57.7 years, and the female-to-male ratio was 3:1, consistent with previously reported series.
Our findings confirm the heterogeneity of PBMAH. Overt CS was relatively rare, occurring in only 6.8% of cases, whereas the majority of patients (58.7%) were classified as having MACS. This observation is consistent with previous studies reporting a predominance of subclinical disease in PBMAH cohorts. However, the clinical impact of MACS should not be underestimated. Even mild hypercortisolism in patients with MACS was associated with cardiometabolic comorbidities, including hypertension, diabetes mellitus, hyperlipidemia, and osteoporosis.18,19 In our cohort, approximately 70% of patients with MACS demonstrated at least one of these comorbidities, emphasizing the importance of careful clinical monitoring.
Differential diagnosis from bilateral adrenal adenomas and micronodular adrenal hyperplasia is essential. CT and MRI remain the primary imaging modalities, with bilateral nodules >1 cm supporting the diagnosis of macronodular disease. When available, adrenal volume measurement and nodule size quantification can further improve diagnostic accuracy. Nodule size and bilaterality also influence the diagnostic evaluation of the disease.17,18 2-(fluorine 18) fluoro-2-dexoxy-D glucose (18 F-FDG) positron emission tomography/CT (PET/CT) is another imaging modality used in the evaluation of PBMAH. Although a standardized uptake value >3.1 suggests a malignant lesion, standardized uptake values in PBMAH are typically low.1,20 We performed PET/CT for preoperative lateralization in one patient, which demonstrated similarly low FDG uptake on both sides.
Management strategies for PBMAH should be individualized. The European Society of Endocrinology guidelines recommend surgery for patients with post-DST cortisol >140 nmol/L or those with progressive hypercortisolism, whereas conservative follow-up is considered for those with normal or mildly elevated cortisol levels without comorbidities. 4 The consensus on treatment decision-making for PBMAH states that patients with normal UFC levels, normal ACTH levels, cortisol <140 nmol/L following 1-mg overnight DST, and those with no hypercortisolism-related comorbidities may be managed with monitoring, follow-up, and steroid suppressor medical therapy.21,22 The European Society of Endocrinology guidelines further suggest surgical treatment for patients with plasma cortisol levels >140 nmol/L after a 1-mg DST.4,11 Surgery may also be considered in patients with disease progression during follow-up and in those without improvement under medical therapy.22,23 In our cohort, five patients underwent unilateral laparoscopic adrenalectomy (UA), achieving complete biochemical remission and resolution of clinical symptoms without adrenal insufficiency during follow-up. These findings support UA as a safe and effective alternative to bilateral adrenalectomy, particularly in patients with overt CS or severe MACS, as it reduces perioperative morbidity and avoids lifelong steroid dependence.
Bilateral adrenalectomy is an effective strategy for controlling hypercortisolism and has been used in patients with overt CS. However, it results in permanent adrenal insufficiency, requiring lifelong glucocorticoid and mineralocorticoid replacement therapy, which is associated with reduced quality of life. Furthermore, the procedure carries a mortality rate of 6%–8%, underscoring the significant perioperative risks of bilateral adrenalectomy. 13 In contrast, recent studies have shown that unilateral adrenalectomy provides a safer alternative, achieving remission in approximately 60% of patients with CS and minimize surgical complications and perioperative morbidity. 14
A review from Canada evaluated 117 patients who underwent UA across 23 studies, with individual study sample sizes ranging from 1 to 25 patients. The initial remission rate was 93%, and biochemical relapse occurred in 18 patients (15%). Of them, 16 patients subsequently underwent contralateral adrenalectomy at a mean of 72 months after the initial surgery. 22
Some authors have proposed subtotal adrenalectomy as a conservative surgical option for patients with PBMAH, involving total adrenalectomy on one side and preservation of approximately one-third of the contralateral adrenal gland. When UA was compared with adrenal-sparing surgery, approximately half of the patients (48%) who underwent adrenal-sparing procedures required long-term corticosteroid replacement therapy (p = 0.02). 14
When unilateral adrenalectomy is selected, determining which adrenal gland to remove can be challenging. The European Society of Endocrinology and Endocrine Society guidelines recommend removal of the gland responsible for cortisol excess and, in selected cases, the larger adrenal gland in patients with PBMAH. 21
Selection of the adrenal gland for unilateral resection is based on imaging findings, biochemical lateralization, and clinical severity.13,23,24 The limited availability of NP-59 scintigraphy and adrenal venous sampling has restricted their routine use; however, these modalities may be valuable in selected cases.25,26
In our cohort, five patients (8.6%) underwent UA, all of whom achieved biochemical remission without relapse or adrenal insufficiency during follow-up. Patients also showed improvement in hypercortisolism-related symptoms. These findings support the growing consensus that unilateral adrenalectomy is a viable alternative to bilateral adrenalectomy in patients with overt CS or severe MACS. 13
The limitations of this study include its retrospective design, relatively short follow-up duration (median, 14 months), and restriction of genetic analysis to ARMC5. Furthermore, KDM1A was not included in the genetic panel; therefore, patients could not be evaluated for KDM1A-associated disease or food-dependent CS. Future studies incorporating broader NGS panels, whole-exome or whole-genome sequencing, and longer follow-up periods may provide further insight into the pathogenesis of PBMAH, genotype–phenotype correlations, and optimal management strategies.
Conclusion
PBMAH is a rare but clinically significant cause of ACTH-independent CS. Most patients in this cohort presented with MACS, underscoring the importance of careful evaluation of cardiometabolic comorbidities even in the absence of overt CS. Genetic testing for ARMC5 mutations is strongly recommended for diagnostic confirmation, familial risk assessment, and cascade screening of relatives. Identified families were offered genetic counseling to support surveillance, early diagnosis, and informed clinical decision-making. Future studies should include evaluation of additional candidate genes (e.g. KDM1A), investigation of somatic mutations, and integration of advanced imaging modalities and NGS techniques to refine individualized management strategies and improve long-term outcomes. Unilateral adrenalectomy is a safe and effective treatment option for patients with overt CS or severe MACS, whereas conservative management is appropriate for nonfunctional or mild cases.
Footnotes
Acknowledgments
Not applicable.
Author contributions
Emine Özlem Gür: Design of the study, evaluate data, and writing the manuscript. Serkan Karaıslı: Collecting the data and writing the manuscript. Ümit Çavdar: Evaluate data and writing the manuscript. Aslı Subaşıoğlu: Genetic testing of the patients, evaluate data, and writing the manuscript. Özlem Eren: Collecting data and data analysis. Selda Gücek Hacıyanlı: Collecting data. Barış Önder Pamuk: Collecting data and data analysis. Mehmet Sercan Ertürk: Collecting data. Mehmet Hacıyanlı: Writing manuscript and critical evaluating.
Consent
Written consent was obtained from the patients who were operated and the patient whose photo was used. It was not appropriate to obtain consent for the other patients who were not operated on.
Data availability statement
Data available on request from the authors.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical consideration
The study adhered to the Declaration of Helsinki (2024 update), and approval was granted by our institutional ethics committee (Approval No: 150/2025). All patient data were fully deidentified.
Funding
All of the authors declare that there is no financial disclosure in connection with this paper.
Supplemental material
Supplemental material for this article is available online.