
Abstract
Objective
Although biologic therapies targeting cytokines have revolutionized the treatment of rheumatoid arthritis, immune cell surface markers remain underexplored as therapeutic targets. We combined Mendelian randomization with clinical evidence to identify causal immune cell phenotypes as potential causal contributors to rheumatoid arthritis and as candidate drugs and therapeutic targets.
Methods
We analyzed genome-wide association study summary statistics from rheumatoid arthritis cohorts (discovery: 12,555 cases/240,862 controls; replication: 14,361 cases/43,923 controls) as well as 731 immune cell traits. The primary analysis used inverse-variance weighted Mendelian randomization. Clinical trial evidence was further used to explore the therapeutic potential of identified targets. Associations with p < 0.05 were considered nominally significant, while Bonferroni correction was applied to determine statistical significance (p < 6.83 × 10−5).
Results
Analysis of the discovery cohort identified 92 nominally associated immune phenotypes, with 10 surviving multiple testing correction. Replication analysis showed 88 nominal associations, with 4 passing the correction. Meta-analysis revealed suggestive evidence for 17 phenotypes. Five immune markers (CD28, CD27, CX3CR1, CD3, and human leukocyte antigen (HLA)-D-related (DR)) emerged as potential diagnostic and therapeutic targets, supported by clinical trial evidence.
Conclusions
This study identified immune biomarkers as potential diagnostic and therapeutic targets for rheumatoid arthritis, providing a framework for prioritizing targetable pathways beyond cytokine blockade and offering new therapeutic avenues.
Keywords
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disorder characterized by persistent inflammatory arthritis, leading to progressive joint damage and systemic manifestations. 1 The global age-standardized prevalence rate has been reported to be 224.25 per 100,000 population. 2 Early detection and timely intervention are critical for preventing irreversible joint destruction, which may occur in up to 90% of affected individuals without appropriate management.3–5 According to the 2010 American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) classification criteria, RA diagnosis primarily relies on serological markers such as rheumatoid factor (RF) and anti-citrullinated protein antibody (ACPA).6–8 Generally, ACPA demonstrates higher sensitivity and specificity than RF, with reported diagnostic performance showing a sensitivity of 54% and a specificity of 96%. 9 However, it is important to note that a subset of patients with RA may still test negative for these autoantibodies.10–12 Therefore, the identification of additional reliable cellular biomarkers is essential for improving the diagnosis of RA.
Emerging evidence suggests that RA involves a complex interplay of immune cells and associated molecules, including autoreactive CD4+ T cells, pathogenic B cells, macrophages, inflammatory cytokines, chemokines, and autoantibodies. 13 The chronic inflammatory state and synovial swelling in RA are driven by functional abnormalities across multiple intra-articular cell types, particularly T and B lymphocytes, osteoclasts, and synoviocytes. This persistent inflammatory state leads to sustained joint pain, accelerated cartilage degradation, and progressive bone erosion. 4 Immunomodulatory strategies, such as T and B cell depletion therapies and the application of tolerogenic dendritic cells (DCs), have demonstrated potential as therapeutic approaches for RA. However, a substantial body of the existing knowledge is derived from observational studies, which are inherently limited by reverse causation and confounding factors. Consequently, it remains challenging to determine whether immune cell alterations represent causes or consequences of RA, thereby hindering the identification of pathogenic immune cells and causal biomarkers for early diagnosis and therapeutic targeting.8,14
To address these limitations, high-quality evidence derived from robust causal inference methods is required. Mendelian randomization (MR) is an epidemiological approach that uses genetic variants as instrumental variables (IVs) to infer causal relationships between an exposure and an outcome. By leveraging the random allocation of alleles during meiosis, MR mitigates confounding and reverse causation biases that are common in observational studies.15,16 In this study, we employed a two-sample MR framework using genome-wide association study (GWAS) summary data for immune cell traits and RA to systematically evaluate the causal effects of immune cell phenotypes on RA pathogenesis. Furthermore, we integrated MR-derived causal evidence with existing observational and clinical trial data to assess the translational potential of surface markers on identified immune cells as causal contributors and therapeutic targets in RA.
Methods
Study design and data source
Study design. This study is a unidirectional MR analysis based on summary-level data, and this report was prepared in accordance with the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE)-MR reporting guidelines (please refer to the Supplementary Materials for the STROBE-MR report). The framework employed in this study is shown in Figure 1. We utilized GWAS summary statistics for immune cells as exposures and GWAS summary statistics for RA as the outcome. To enhance statistical power, we conducted a discovery analysis using the FinnGen BioBank GWAS dataset, which comprises 253,417 participants of Finnish ancestry, followed by a replication analysis using the IEU open GWAS dataset, which includes 58,284 participants of European ancestry. Finally, the results from both analyses were integrated using a meta-analysis approach. 17

Framework of MR analysis in this study. cDC: conventional dendritic cell; kb: kilobase; MFI: median fluorescence intensities; MR: Mendelian randomization; PRESSO: pleiotropy residual sum and outlier test; RA: rheumatoid arthritis; SNP: single-nucleotide polymorphisms; TBNK: T cell, B cell, natural killer cell; Treg: regulatory T cell.
Data source. Regarding exposure datasets, summary statistics for a total of 731 immune cell traits were obtained from the GWAS catalog (accession numbers: GCST0001391 to GCST0002121). 18 These traits were assessed in a population-based cohort of 3757 healthy Sardinian individuals. The dataset encompassed 118 absolute cell counts (AC), 389 median fluorescence intensities (MFI) reflecting surface antigen expression levels, 32 morphological parameters, and 192 relative cell counts. Genotyping was conducted using four Illumina arrays—OmniExpress, ImmunoChip, Cardio-MetaboChip, and ExomeChip. Genome-wide imputation and association analyses were performed using a Sardinian-specific reference panel comprising 3514 individuals who had undergone sequencing. Ultimately, association testing was conducted for approximately 22 million high-quality genetic variants, adjusting for age, age squared, and sex. 19 Comprehensive information, including sex distribution, age at diagnosis, living environment, and economic status, is available in the primary publications. 19
Regarding outcome datasets, GWAS summary statistics for the discovery analysis of RA were obtained from the R12 release of the FinnGen GWAS database (https://r12.finngen.fi/). 20 This dataset comprised 12,555 RA cases and 240,862 controls—all of European ancestry. GWAS summary statistics for the replication cohort were retrieved from the IEU Open GWAS Project (https://gwas.mrcieu.ac.uk/; GWAS ID: ieu-a-832). 21 This dataset included 14,361 RA cases and 43,923 controls—also of European ancestry. Comprehensive information on these two datasets, including sex distribution, age at diagnosis, living environment, and economic status, is available in primary publications. 21 We used independent discovery and replication cohorts, followed by a meta-analysis, to overcome the limitations of single-cohort analyses and ensure robust, generalizable causal inferences. Integrating both cohorts enhanced statistical power, reduced random variation, and yielded more precise estimates of the causal effects of immune cell traits on RA.
Assumptions. This study employed a unidirectional MR design to assess the causal effects of immune cell phenotypes on RA risk. The MR analysis was based on three key assumptions 22 : (a) the genetic variant is significantly associated with immune cells; (b) the genetic variant is not associated with potential confounders between exposure and outcome; and (c) the genetic variant is not directly related to RA (Figure 2).

MR model. GW: genome wide; IV: instrumental variable; IVW: inverse-variance weighted; MR: Mendelian randomization; SNP: single-nucleotide polymorphisms; WM: weighted median.
Selection criteria of IVs. To identify genetic instruments for immune cell traits, we selected single-nucleotide polymorphisms (SNPs) based on a significance threshold of p < 1 × 10−5, as the number of variants meeting the conventional genome-wide significance threshold (p < 5 × 10−8) was limited. This approach has been recommended in previous studies. 23 To ensure genetic independence among instruments, we performed linkage disequilibrium (LD) clumping using an LD threshold of R2 <0.001 within a 10,000 kb window, based on the 1000 Genomes Project reference panel. 24 We also excluded palindromic SNPs and those with a minor allele frequency below 0.01.
The strength of each IV was assessed using the F-statistic, calculated as follows:
F = R2 (N − K − 1)/[K (1 − R2)], where R2 is the proportion of variance explained by the SNP, N is the sample size, and K is the number of SNPs. Subsequently, SNPs with an F-statistic below 10 were considered weak instruments and excluded from subsequent analyses. 25
Statistical analysis
The primary MR analysis was conducted using the inverse-variance weighted (IVW) method, 26 as it provides the most precise causal estimates and the highest statistical power in the absence of horizontal pleiotropy. 27 To ensure the robustness of the findings, additional MR methods were applied, including the weighted median, weighted mode, 28 and MR-Egger regression. 29 A result was considered statistically significant only if the IVW method demonstrated a significant association and all sensitivity analyses (MR-Egger regression, weighted median method, and weighted mode) produced effect estimates consistent in direction with the IVW estimate. To evaluate potential horizontal pleiotropy (a genetic phenomenon in which a single genetic variant affects multiple distinct traits), we implemented multiple strategies, including the MR-Egger intercept test. Furthermore, the leave-one-out analysis was performed to identify influential SNPs; outliers were removed, and the IVW analysis was repeated thereafter. Funnel plots were used to assess potential systematic bias or pleiotropy. Symmetry in the funnel plot indicates the absence of such bias. Heterogeneity among SNPs was assessed using Cochran’s Q test, with a p-value of less than 0.05 indicating significant heterogeneity.
The aforementioned MR analyses were applied to both the discovery and replication cohorts. A meta-analysis of the results from these two cohorts was conducted using the META package.30,31 The choice of the effect model was determined by the degree of heterogeneity observed between the results of the discovery and replication cohorts. In cases of low heterogeneity (I2 ≤ 50%), a fixed-effects model was used for the meta-analysis. When substantial heterogeneity was observed (I2 > 50%), a random-effects model was applied. We used Venn diagrams to identify immunophenotypes overlapping between the discovery and the replication RA cohorts.
Nominal or suggestive significance was defined as p < 0.05. To correct for multiple comparisons across the 731 immune traits, Bonferroni correction was applied, yielding a significance threshold of p < 6.83 × 10−5 (0.05/731).32,33 Statistical significance was defined using a threshold of p < 6.83 × 10−5. All outcomes are reported as odds ratios (ORs) with corresponding 95% confidence intervals (CIs).
All statistical analyses were conducted using R software (version 4.3.1), TwoSampleMR package, and META package. The results of the IVW MR analysis were presented as scatter plots, while the corresponding weighted estimates were displayed as forest plots.
Druggability of targets
We assessed the druggability of immune markers associated with the identified immunophenotypes by referencing the International Clinical Trials Registry Platform (ICTRP) and ClinicalTrials databases. All therapeutic modalities—including small-molecule inhibitors, monoclonal antibodies, and other biologics—were evaluated according to their developmental stage (e.g. preclinical or clinical phases) and the supporting evidence for their efficacy in RA or other autoimmune disease.34–39
Ethical considerations
All datasets used in this study were obtained from publicly accessible online sources, with no original data collection involved. Thus, ethics committee approval was not required.
Patient and public involvement
This study did not involve individual-level data.
Results
Causal impact of genetically predicted immunophenotypes on RA
Discovery cohort. As shown in Figure 3 and Table S1, a total of 92 genetically predicted immunophenotypes in the discovery cohort showed suggestive associations with RA (p < 0.05). Among them, 10 traits reached statistical significance after Bonferroni correction (p < 6.83 × 10−5) (Figure 3 and Table S1). Notably, seven immunophenotypes were identified as potential risk factors, including human leukocyte antigen (HLA)-D-related (DR) expression on plasmacytoid DCs (pDCs) (OR = 1.48; 95% CI: 1.31–1.68; p = 4.47 × 10−10), HLA-DR on CD14+CD16− monocytes (OR = 1.60; 95% CI: 1.38–1.85; p = 5.33 × 10−10), HLA-DR on CD14+ monocytes (OR = 1.60; 95% CI: 1.37–1.87; p = 2.29 × 10−9), HLA-DR on CD33+HLA-DR+ cells (OR = 1.53; 95% CI: 1.29–1.80; p = 5.72 × 10−7), HLA-DR on myeloid DCs (OR = 1.65; 95% CI: 1.31–2.09; p = 2.52 × 10−5), HLA-DR on total DCs (OR = 1.45; 95% CI: 1.22–1.73; p = 3.11 × 10−5), and HLA-DR on CD33br HLA-DR+CD14dim cells (OR = 1.41; 95% CI: 1.22–1.64; p = 5.53 × 10−6), all of which were significantly associated with an increased risk of RA. Additional MR methods, including the weighted median, weighted mode, and MR-Egger regression, confirmed the robustness of these results.

Mendelian randomization analysis of inverse-variance weighted method results in the discovery cohort. %DC: percentage of dendritic cells; AC: absolute count; CD: cluster of differentiation; DC: dendritic cell; FSC-A: forward scatter-area; HLA-DR: human leukocyte antigen-D-related; HVEM: herpesvirus entry mediator; MFI: median fluorescence intensities; mDC: myeloid dendritic cell; OR: odds ratio; pDC: plasmacytoid dendritic cell.
In contrast, three immunophenotypes exhibited protective effects against RA. Within the TBNK (T, B, and natural killer (NK) cell) panel, side scatter-area (SSC-A) of HLA-DR+ NK cells was significantly associated with a decreased risk of RA (OR = 0.90; 95% CI: 0.86–0.94; p = 9.03 × 10−6). In the monocyte panel, CD40 expression on CD14+CD16− monocytes (OR = 0.91; 95% CI: 0.88–0.95; p = 1.07 × 10−5) and on total monocytes (OR = 0.93; 95% CI: 0.89–0.96; p = 5.79 × 10−5) also demonstrated significant protective effects. Additional MR methods, including weighted median, weighted mode, and MR-Egger regression, confirmed the robustness of these results (Table S2).
These results were further supported by leave-one-out analyses and funnel plots, confirming the robustness and consistency of the findings (Figures S1 and S2).
Replication cohort
In the replication cohort, 88 genetically predicted immunophenotypes showed suggestive associations with RA (p < 0.05), among which 4 immunophenotypes reached statistical significance after Bonferroni correction (p < 6.83 × 10−5) (Figure 4 and Table S3). These four significant immunophenotype–RA pairs were as follows: CD25 expression on IgD-CD38− B cells was positively associated with RA (OR = 1.25; 95% CI: 1.16–1.35; p = 8.05 × 10−9); IgD-CD27−% B cells showed a positive causal relationship with RA (OR = 1.32; 95% CI: 1.17–1.50; p = 9.08 × 10−6); in the conventional DC (cDC) panel, a significant positive association was identified for CD62L−CD86+ myeloid DC %DC (OR = 1.12; 95% CI: 1.06–1.18; p = 4.63 × 10−5); and CD11b expression on CD33br HLA-DR+ CD14dim cells was identified as a protective factor against RA (OR = 0.87; 95% CI: 0.82–0.93; p = 2.83 × 10−5). Additional MR methods confirmed the robustness of these results (Table S4).

Mendelian randomization analysis of inverse-variance weighted method results in the replication cohort. %DC: percentage of dendritic cells; AC: absolute count; CD: cluster of differentiation; CX3CR1: C-X3-C motif chemokine receptor 1; DC: dendritic cell; FSC-A: forward scatter-area; HLA-DR: human leukocyte antigen-D-related; HVEM: herpesvirus entry mediator; MFI: median fluorescence intensities; mDC: myeloid dendritic cell; OR: odds ratio; pDC: plasmacytoid dendritic cell.
These results were further supported by sensitivity analyses, including the leave-one-out analyses and funnel plots, confirming the robustness and consistency of the findings (Figures S3 and S4).
Meta‐analysis
We examined overlapping immunophenotypes with nominally significant MR results (p < 0.05) between the discovery and replication cohorts (Table S5). A Venn diagram revealed that 26 immunophenotypes were shared between the two cohorts (Figure 5(a)). Subsequently, we performed a meta-analysis of these overlapping immunophenotypes. The meta-analysis identified 17 immunophenotypes associated with RA risk. Among them, 13 immunophenotypes were risk factors for RA, including HLA-DR on pDCs (cDC panel; OR = 1.49; 95% CI =1.33–1.68; p < 0.0001), DC absolute count (cDC panel; OR = 1.07; 95% CI = 1.02–1.12; p = 0.0022), CD62L−CD86+ myeloid DC %DC (cDC panel; OR = 1.09; 95% CI =1.05–1.14; p < 0.0001), CD62L−CD86+ myeloid DC absolute count (cDC panel; OR = 1.09; 95% CI = 1.05–1.13; p < 0.0001), myeloid DC absolute count (cDC panel; OR = 1.09; 95% CI = 1.05–1.13; p < 0.0001), HLA-DR on CD33−HLA-DR+ (myeloid cell panel; OR = 1.39; 95% CI = 1.14–1.69; p = 0.0011), CD33dim HLA-DR+ CD11b−% CD33dim HLA-DR+ (myeloid cell panel; OR = 1.07; 95% CI = 1.00–1.14; p = 0.0490), naive CD4+ T cell absolute count (maturation stages of T cell panel; OR = 1.48; 95% CI = 1.13–1.94; p = 0.0050). CD28 on CD28+ CD45RA+ CD8+ T cell (Treg panel; OR = 1.11; 95% CI =1.06–1.16; P < 0.0001), IgD-CD27-B cell % B cell (B cell panel; OR = 1.22; 95% CI = 1.04–1.41; p = 0.0120), CX3CR1 on CD14−CD16+ monocyte (monocyte panel; OR = 1.12; 95% CI = 1.05–1.19; p = 0.0007) HLA-DR on CD14−CD16− (monocyte panel; OR = 1.17; 95% CI = 1.03–1.34; p = 0.0179), CX3CR1 on CD14+ CD16− monocyte (monocyte panel; OR = 1.06; 95% CI = 1.01–1.10; p = 0.0083), and four immunophenotypes are protective factors for RA:CD27 on IgD+ CD38-unswitched memory B cell (B cell panel; OR = 0.94; 95%CI = 0.90–0.97; p = 0.0007), CD14−CD16+ monocyte absolute count (monocyte panel; OR = 0.93; 95% CI = 0.89–0.98; p = 0.0042), CD3 on CD39+ activated CD4 regulatory T cell (Treg panel; OR = 0.94; 95% CI = 0.09–0.97; p = 0.0002), CD3 on activated CD4 regulatory T cell (Treg panel; OR = 0.95; 95% CI = 0.92–0.98; p = 0.0012) (Figure 5(b)).
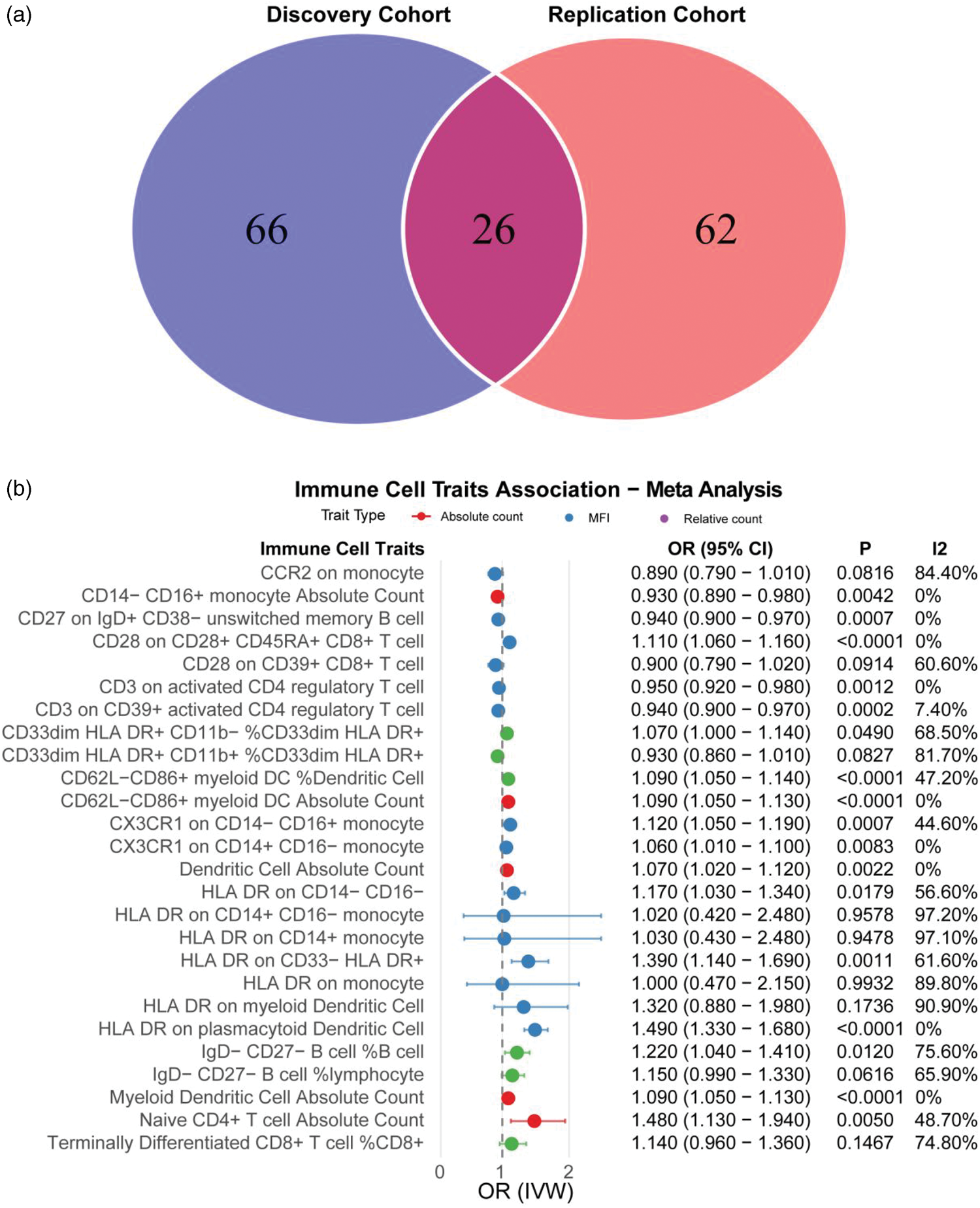
Venn diagram of the discovery cohort and replication cohort (a) and forest plot of results of meta‐analysis for the overlapped portion (b). %DC: percentage of dendritic cells; AC: absolute count; cDC: conventional dendritic cell; CX3CR1: C-X3-C motif chemokine receptor 1; DC: dendritic cell; HLA-DR: human leukocyte antigen-D-related; I2: heterogeneity index; MFI: median fluorescence intensities; OR: odds ratio.
Drug actionability
Among the 17 immunophenotypes identified through meta-analysis, we further evaluated drugs targeting their surface markers using the ICTRP and ClinicalTrials databases to assess their potential efficacy in RA. Surface markers that had been validated in clinical trials were defined as therapeutic targets. Based on this criterion, we prioritized five immunophenotypic markers (29%)—HLA-DR, CD28, CD27, CX3CR1, and CD3—as potential early immune biomarkers and therapeutic targets (Table 1). Notably, genetically predicted increased expression levels of three (18%) markers were associated with an elevated risk of RA. Several therapeutic agents targeting these surface markers have been developed or are under clinical investigation, including monoclonal antibodies, small-molecule inhibitors, and immunomodulatory fusion proteins.40–43 Among these agents, tofacitinib, which targets the CD3 signaling pathway as a Janus kinase (JAK) inhibitor, has been approved by the Food and Drug Administration (FDA) as a first-line targeted therapy for RA. 44 In addition, abatacept, a CTLA-4-Ig fusion protein that modulates CD28 signaling, has been approved for the treatment of RA, particularly in patients who are intolerant or unresponsive to methotrexate (MTX). 45 Our analysis further suggests that CX3CR1 and CD27 may represent attractive therapeutic targets. KAND567, a small-molecule inhibitor targeting CX3CR1, is currently being evaluated in clinical trials for multiple sclerosis and coronavirus disease 2019.46,47 Meanwhile, varlilumab, a monoclonal antibody against CD27, is primarily undergoing clinical evaluation in solid tumor settings (Table S6). 48
Genetically predicted increased levels of immune cell surface markers and RA risk.
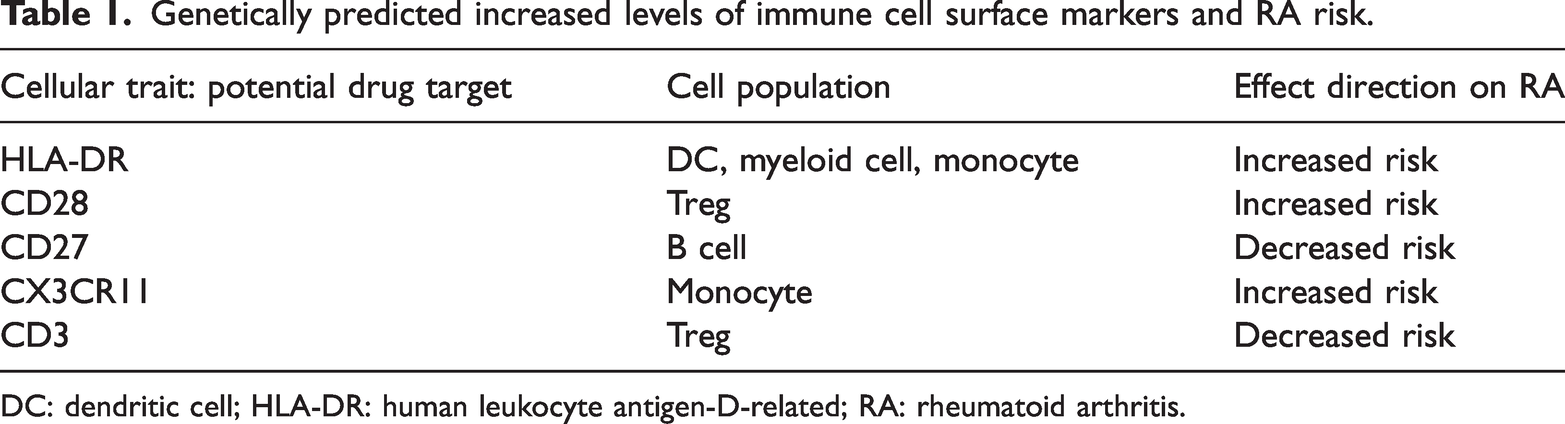
DC: dendritic cell; HLA-DR: human leukocyte antigen-D-related; RA: rheumatoid arthritis.
Discussion
Overall, our MR analysis leveraged large-scale, publicly available GWAS datasets to systematically investigate the associations between immune phenotypes and RA risk. To enhance the robustness of our findings, we performed a comprehensive meta-analysis integrating results from both the discovery and replication cohorts. The meta-analysis identified 13 immune phenotypes (HLA-DR on pDCs; myeloid DC absolute count; CD28 on CD28+ CD45RA+ CD8+ T cell; CD62L−CD86+ myeloid DC absolute count; lgD-CD27− B cell %B cell; CX3CR1 on CD14−CD16+ monocyte; CD33dim HLA-DR+ CD11b−%CD33dim HLA-DR+; DC absolute count; HLA-DR on CD14.CD16−; naive CD4+T cell absolute count, CD62L−CD86+ myeloid DC %DC; and CX3CR1 on CD14+ CD16− monocyte) as significant risk factors for RA development and four immune phenotypes (CD27 on IgD+CD38− unswitched memory B cell; CD14−CD16+ monocyte absolute count; CD3 on CD39+ activated CD4 regulatory T cell; and CD3 on activated CD4 regulatory T cell) as protective factors. Importantly, we further prioritized five immune cell surface markers as promising candidates as potential causal contributors and therapeutic targets in RA management. These findings provide novel insights into the immunopathological mechanisms of RA and highlight potential avenues for targeted intervention strategies.
Our study showed that elevated levels of five immunophenotypes from the cDC group—HLA-DR on pDCs, myeloid DC absolute count, CD62L−CD86+ myeloid DC absolute count, DC absolute count, and CD62L−CD86+ myeloid DC %DC—were associated with an increased risk of RA. Dendritic cells are key specialized antigen-presenting cells that link innate and acquired immune responses by initiating and driving the differentiation of naive cells into effector cells, and their interactions are essential for the generation of autoimmune responses in RA. 49 Myeloid DCs may play an important role in the pathogenesis of RA due to their efficient antigen-presenting capacity. 50
According to our findings, RA was positively associated with elevated levels of HLA-DR on CD33−HLA-DR+ as well as CD33dim HLA-DR+ CD11b− immunophenotypes in the myeloid cell group. These findings suggest that the activation status of specific myeloid cell subsets may play a pivotal role in RA development. The increased expression of HLA-DR on CD33− and CD33 dim cells indicates enhanced antigen-presenting capacity, which may contribute to the persistent activation of autoreactive T cells in RA. 13 CD33 is a myeloid marker typically expressed on immature myeloid cells, and its dim or negative expression reflects a more differentiated or activated phenotype. 51 The coexpression of high levels of HLA-DR, a key major histocompatibility complex (MHC) class II molecule, further supports the notion that these myeloid cells exist in an activated immunostimulatory state. 52 Moreover, the absence of CD11b, a marker involved in leukocyte adhesion and migration, may indicate altered trafficking or functional specialization of these cells within the inflammatory microenvironment of RA. 20 Collectively, these immunophenotypic characteristics point toward aberrant activation of myeloid antigen-presenting cells, which may sustain chronic inflammation and promote autoimmunity in RA. 53 Further functional studies are warranted to clarify the roles of these specific myeloid subsets in RA pathogenesis and to assess their potential as biomarkers or therapeutic targets.
In the monocyte group, three immunophenotypes—CX3CR1 on CD14−CD16+ monocyte, HLA-DR on CD14−CD16−, and CX3CR1 on CD14+CD16− monocyte, were positively associated with RA risk. In contrast, CD14−CD16+ monocyte absolute count showed a negative correlation. Monocyte subpopulations can be categorized according to phenotypic markers as classic (CD14+CD16−), intermediate (CD14+CD16+), and atypical (CD14−CD16+) subsets. Monocytes express the chemokine receptors CX3CR1 and CCR2, which interact with the chemokine ligands CX3CL1 (fractalkine) and CCL2 (MCP-1) secreted by synoviocytes and fibroblast-like cells. These interactions facilitate the recruitment and migration of monocytes to the RA-affected synovium. Furthermore, monocyte activation leads to the expression of antigens, such as CD14 and CD16, which subsequently influence RA progression. 54 A clinical study showed that circulating CD16+ monocytes exhibited higher levels of CX3CR1 expression than CD16− monocytes in patients with RA and healthy individuals. 55 At the same time, another study’s results revealed that CD14+CD16− monocytes are the primary source of osteoclasts, 56 indicating that increased levels of this subset may be positively associated with RA risk. In addition, a clinical trial showed that patients with RA who were nonresponders exhibited increased numbers of monocytes CD14+lowCD16+ (atypical type) subsets after 3 months of adalimumab plus MTX treatment, which remained significantly elevated at 6 months. 57 These findings provide supportive clinical evidence for our research.
In the maturation stage group of T cells, the naive CD4+ T cell absolute count was positively associated with the increased risk of RA. Upon antigenic stimulation, naive CD4+ T cells proliferate and differentiate into distinct effector subpopulations (Th17 cells), which are involved in mediating autoimmune diseases. Studies of the Th17 T cell lineage suggest a broader involvement of T cells in RA pathogenesis. 58 An animal study showed that intra-articular injection of IL-17 in mice induced changes similar to those seen in RA. 59
In the Treg group, two immunophenotypes—CD3 on activated CD4 regulatory T cell and CD3 on CD39+ activated CD4 regulatory T cell—exhibited protective effects against RA occurrence, whereas CD28 on CD28+ CD45RA+ CD8+ T cell indicated was associated with RA risk. However, a previous study showed that CD4+CD3− cells are enriched in patients with RA, suggesting a potential link to chronic inflammatory disease. 60 Another study suggested that among human CD4+ T cells at inflammatory sites, CD39 is highly expressed in two populations—one regulatory and the other with a memory phenotype—and contributes to the progression of various autoimmune diseases through different mechanisms. 61 In addition, an increased frequency of CD8+CD28− cells has been observed in individuals affected by early-stage RA and was associated with the duration of the illness. 62 CD28 encodes a negative regulator of DC/T cell interactions and has been associated with RA pathogenesis. 63 These findings suggest that these cells might potentially contribute to the initial progression of the disease
Our findings also demonstrated a positive association between IgD-CD27− B cell %B cell and the risk of RA in the B cell group. Conversely, CD27 on IgD+ CD38− unswitched memory B cells showed a negative correlation. Single-cell sequencing analyses showed that the proportion of double-negative (CD27−IGD−) B cells was significantly higher in the synovium of patients with RA than in the peripheral blood. 64 Similarly, a clinical study found that double-negative (IgD−CD27−) levels were significantly higher in untreated patients with early RA (disease duration <1 year) and in patients with diagnosed RA (treated with MTX and before initiating anti-TNF or tocilizumab therapy) compared to controls, and returned to normal levels after the corresponding treatment. 65 In contrast to long-standing RA, the pathogenesis is characterized by a lower number of plasmablasts and double-negative memory B cells compared to established RA, and CD38 may be a potential target for disease blockade in RA.66,67
Our study identified several immunophenotypic markers with potential diagnostic and therapeutic values in RA. Among these, CX3CR1 and CD28 emerged as novel druggable targets, supported by genetic evidence linking their increased expression to elevated RA risk. The feasibility of these targets is further supported by existing pharmacological interventions, either approved or under clinical investigation, that modulate their associated pathways. Notably, JAK inhibitors (e.g. tofacitinib) and CTLA-4-Ig fusion proteins (e.g. abatacept)—both targeting pathways related to our identified markers (CD3 and CD28)—have demonstrated clinical efficacy in RA.68,69 These observations suggest that our findings align with established therapeutic mechanisms while also proposing novel avenues for intervention. Specifically, CX3CR1, a chemokine receptor involved in leukocyte adhesion and migration, is currently being explored as a target in inflammatory and autoimmune conditions. The small-molecule inhibitor KAND567, which blocks CX3CR1, is under clinical evaluation for multiple sclerosis and coronavirus disease 2019, highlighting its potential applicability to RA. 70 Similarly, CD27, a costimulatory molecule critical for T cell activation, could be modulated by varlilumab, an anti-CD27 monoclonal antibody currently under investigation in oncology but with plausible translational potential in autoimmune diseases. 48 These findings underscore the importance of repurposing existing drugs or advancing targeted biologics for the treatment of RA. Given that a subset of patients remains refractory to conventional therapies such as MTX or TNF inhibitors, our results provide a strong rationale for exploring alternative mechanisms, particularly those involving CX3CR1 and CD27. Future studies should validate these targets in preclinical RA models and assess whether their inhibition or modulation can ameliorate disease progression. Additionally, biomarker-driven clinical trials could stratify patients based on immunophenotypic profiles to enable personalized therapeutic strategies. 71
Several limitations of this study should be acknowledged. First, to minimize confounding arising from genetic differences across populations, we restricted our analyses to individuals of European ancestry. Although this reduces population stratification bias, it may limit the generalizability of our findings to non-European populations. Second, as a computational study, the diagnostic markers and therapeutic targets identified herein require further experimental and clinical validation to confirm their relevance and clinical utility.
Conclusion
In conclusion, this study elucidated the causal relationships between immune cells and RA by utilizing data from large GWAS and multiple MR analytical methods, an approach that greatly mitigates confounding factors such as environmental and lifestyle influences; the inclusion of a validation cohort increases the stability of the results. The meta-analysis identified 13 immune phenotypes as risk factors and 4 as protective factors for RA, along with 5 key cellular markers, providing valuable insights and guidance for elucidating the pathogenesis, early diagnosis, and therapeutic target development of RA. However, these findings still warrant further validation in experimental and clinical cohorts.
Supplemental Material
sj-xlsx-1-imr-10.1177_03000605251409686 - Supplemental material for Mendelian randomization identifies multiple immune cell surface markers as potential causal contributors and drug targets in rheumatoid arthritis
Supplemental material, sj-xlsx-1-imr-10.1177_03000605251409686 for Mendelian randomization identifies multiple immune cell surface markers as potential causal contributors and drug targets in rheumatoid arthritis by Guangyu Huang, Yunlong Huang, Gui Liao, Kaizhen Xiao, Cun Li and Ronghe Gu in Journal of International Medical Research
Supplemental Material
sj-pdf-2-imr-10.1177_03000605251409686 - Supplemental material for Mendelian randomization identifies multiple immune cell surface markers as potential causal contributors and drug targets in rheumatoid arthritis
Supplemental material, sj-pdf-2-imr-10.1177_03000605251409686 for Mendelian randomization identifies multiple immune cell surface markers as potential causal contributors and drug targets in rheumatoid arthritis by Guangyu Huang, Yunlong Huang, Gui Liao, Kaizhen Xiao, Cun Li and Ronghe Gu in Journal of International Medical Research
Footnotes
Acknowledgments
Not applicable.
Author contributions
Guangyu Huang and Ronghe Gu contributed to the study design; Yunlong Huang, Gui Liao, and Kaizhen Xiao performed conceptualization, statistical analysis, data interpretation, and manuscript drafting and review. Cun Li contributed to manuscript drafting, data analysis, and interpretation. Guangyu Huang and Yunlong Huang have contributed equally in the planning and writing of the manuscript as the first author. All the authors have read and approved the final manuscript.
Consent for publication
Not applicable.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material. Further inquiries, including access to analysis code, can be directed to the corresponding author.
Declaration of conflicting interests
All the authors declare that they have no competing interests.
Ethics considerations
All the datasets in this study were from publicly accessible data online, with no original data collection involved; thus, ethical committee approval was not needed.
Funding
Supported by the Cultivated by Outstanding Young Scientific and Technological Innovation and Entrepreneurship Talents in Nanning City, China, No. RC20210107; and General Project of Guangxi Natural Science Foundation, China, No. 2022JJA141393.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.