
Abstract
Objective
The role of the tumor suppressor p53 apoptosis effector related to PMP-22 (PERP) in breast cancer metastasis remains unclear. This study investigated PERP’s role in metastatic progression, its clinical significance, and the mechanisms underlying its effects.
Methods
PERP expression was assessed in breast cancer cell lines, public datasets, and 142 patient samples using real-time quantitative polymerase chain reaction (RT–qPCR), western blotting, and immunohistochemistry. In vitro migration and invasion assays as well as in vivo metastasis model were performed after PERP overexpression or knockdown in MDA-MB-231 and MCF-7 cells. The roles of activating transcription factor 3 (ATF3) and heat shock protein family A member 6 (HSPA6) were evaluated through small-interfering RNA–mediated modulation, RNA sequencing, western blotting, RT–qPCR, and chromatin immunoprecipitation.
Results
PERP expression was markedly reduced in breast cancer cells and tumor tissues compared with that in normal controls, and low PERP levels were associated with poor prognosis. PERP overexpression suppressed metastasis. Mechanistically, PERP upregulated ATF3 expression, and ATF3 bound to the HSPA6 promoter to activate its transcription. Knockdown of ATF3 or HSPA6 eliminated the antimetastatic effects of PERP.
Conclusions
PERP suppresses breast cancer metastasis by inducing ATF3, which in turn activates HSPA6 transcription. This PERP–ATF3–HSPA6 axis represents a key regulatory pathway and serves as a potential therapeutic target in metastatic breast cancer.
Keywords
Introduction
Breast cancer is one of the most frequently diagnosed malignancies in women. In 2022, breast cancer constituted more than 2.3 million of the nearly 20 million new cancer cases worldwide, making it the second most prevalent cancer worldwide. 1 Despite advances in medical technology that have improved survival rates, metastasis remains a major clinical challenge. Approximately 20%–30% of patients with early-stage breast cancer ultimately develop metastatic disease. 2 Once metastasis occurs, prolonged treatments—often associated with side effects and high costs—are required; however, effective therapies remain limited due to inadequate understanding of the complex mechanisms underlying metastatic progression.
p53 apoptosis effector related to PMP-22 (PERP), a transmembrane protein essential for desmosome assembly, plays a critical role in maintaining tissue integrity and function. PERP knockdown inhibits apoptosis, impairs cell adhesion, increases cytokine production, disrupts Ca2+ homeostasis, and promotes epithelial–mesenchymal transition (EMT)—processes that collectively contribute to cancer progression. 3 Studies have shown that PERP is downregulated in several cancer types, particularly breast cancer, where it acts as a tumor suppressor. 4 However, some reports suggest that PERP may also facilitate tumor progression. 5 The precise role and mechanisms of PERP in tumorigenesis, especially in breast cancer, remain poorly understood.
Given that PERP’s antiproliferative role has been previously established—including our work identifying the E3 adaptor DCAF13 as an upstream regulator that promotes breast cancer proliferation via ubiquitin-mediated suppression of PERP—the present study focuses on PERP-driven motility/invasion and its transcriptional coupling to ATF3/HSPA6. 6 Using clinical breast cancer specimens, in vitro cell lines, and a nude mouse model, we investigated PERP’s role in metastasis and the underlying mechanisms. We found that PERP expression is reduced in breast cancer and is associated with poor distant disease-free survival (D-DFS). PERP overexpression suppressed migration/invasion in vitro and metastasis in vivo while upregulating ATF3 and HSPA6 expression. Conversely, knockdown of ATF3 or HSPA6 attenuated the antimotility effects of PERP, and ATF3 was shown to bind to the HSPA6 promoter. Collectively, these findings support an inhibitory role of PERP in metastasis via the ATF3–HSPA6 axis.
Methods
Bioinformatics analysis
The mRNA expression data were obtained from the Gene Expression Omnibus (GEO) dataset GSE45827, generated using the Affymetrix HG-U133 Plus 2.0 platform. This series comprises 130 primary invasive breast cancer samples (29 luminal A, 30 luminal B, 41 basal, and 30 HER2-positive) and 11 normal breast tissues. 7 Raw/processed data were downloaded from GEO (Series Matrix), and microarray differential expression analysis was performed in R (v4.3.3, R Foundation, https://www.R-project.org) using the limma package (v3.58.1, Bioconductor), with moderated t-statistics with Benjamini–Hochberg false discovery rate (FDR) correction. 8 Results were reported as log2 fold change (log2FC) and FDR-adjusted P-values; unless otherwise specified, significance was defined as FDR < 0.05 and |log2FC| ≥ 1. 9
The PERP overexpression RNA sequencing (RNA-seq) dataset was analyzed in R using the DESeq2 package (v1.42.0, Bioconductor), with size-factor normalization (median-of-ratios), Wald tests, and Benjamini–Hochberg FDR correction. 10 Statistical significance was defined as FDR < 0.05 and |log2FC| ≥ 1.
Transcription factor predictions were obtained from hTFtarget (http://bioinfo.life.hust.edu.cn/hTFtarget), and promoter sequences were retrieved from National Center for Biotechnology Information (NCBI). Predicted transcription factor binding sites were analyzed using JASPAR (https://jaspar.elixir.no/).11,12
Cells and cultures
All experiments were performed using mycoplasma-free cell lines. MCF-10A (CSTR: 19375.09.3101HUMGNHu50; RRID: CVCL_0598), MCF-7 (CSTR: 19375.09.3101HUMTCHu74; RRID: CVCL_0031), MDA-MB-231 (CSTR: 19375.09.3101HUMTCHu227; RRID: CVCL_0062), and SK-BR-3 (CSTR: 19375.09.3101HUMTCHu225; RRID: CVCL_0033) were obtained from the National Collection of Authenticated Cell Cultures (Shanghai, China). Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; L100-500, BDBIO, Hangzhou, China) supplemented with 10% fetal bovine serum (S-FBS-SA-015, Serana, Waltham, MA, USA) and 1% penicillin–streptomycin (BL505A, Biosharp, Hefei, China) at 37°C under 5% CO2. Subculturing was performed at a 1:2 or 1:4 ratio upon reaching 80%–90% confluency, and cells in the logarithmic growth phase were used for experiments.
MDA-MB-231-luciferase cells (BNCC337894, BeNa Culture Collection) were used for bioluminescence experiments (the supplier does not provide an RRID for this derivative; the parent line is MDA-MB-231, RRID: CVCL_0062). Cells were seeded into 6-well plates at a density of 1.2 × 105 cells per well and cultured for 24 h until reaching 70% confluency. The cells were then transduced with viral vectors (vector or PERP) at varying multiplicities of infection (MOI; 10, 20, 30, 40, and 50), with the optimal MOI determined to be 50 based on fluorescence intensity measured 72 h post-infection. Polybrene was added to enhance viral transduction efficiency. Following infection, cells were incubated with 3 µg/mL puromycin for 3 days, followed by incubation with 2 µg/mL puromycin to select for stably transduced cells. After selection, cells were expanded in 10-cm dishes under continuous puromycin selection to maintain stable gene expression.
Transient PERP overexpression was employed for rapid, mechanism-focused in vitro assays. For in vivo experiments, a stable PERP-overexpressing MDA-MB-231 cell line was used to ensure sustained expression throughout the study.
Cell transfection
Cells were seeded into 6-well plates at a density of 2 × 105 cells per well. Transfections were performed using the Lipofectamine 3000 kit (L3000015, Thermo Fisher) according to the manufacturer’s instructions. Two 1.5-mL centrifuge tubes were prepared for transfection. In tube A, 250 µL of Opti-MEM and 5 µL of Lipofectamine 3000 were combined. For small-interfering RNA (siRNA) transfection, 250 µL of Opti-MEM and siRNA (at a final concentration of 50 nM) (Table A1) were added to tube B. For plasmid transfection, 250 µL of Opti-MEM, 2.5 µg of plasmid DNA (General Biol, Chuzhou, China), and 5 µL of P3000 were added to tube B. The contents of tubes A and B were mixed and then added dropwise to the cells, followed by incubation for 48 h.
Association between PERP expression and clinicopathological features of breast cancer.

NST: no special type; ER: estrogen receptor; PR: progesterone receptor; HER2: human epidermal growth factor receptor 2; TNBC: triple-negative breast cancer; PERP: p53 apoptosis effector related to PMP-22.
Bold values indicate statistical significance (p ≤ 0.05).
Scratch assay
Cells were seeded into 24-well plates at a density of 3 × 105 cells per well. Upon reaching near confluence, a micropipette was used to create a scratch in the center of each well, after which the plate was transferred to an incubator for further incubation. Images were acquired at 0, 24, and 48 h using a 10× objective (scale bar, 200 μm), and each well was imaged at three pre-marked positions. The cell migration rate at N hours was calculated as follows: (scratch width at 0 h − scratch width at N hours) / scratch width at 0 h. Scratch width was measured via ImageJ at the same three positions per well and averaged. Data were obtained from three independent experiments (n = 3).
Transwell assay
Cell invasion was evaluated using Transwell chambers (Millipore, Billerica, MA, USA) coated with Matrigel. A total of 1 × 105 cells were seeded into the upper chamber, and 600 μL of medium containing 10% FBS was added to the lower chamber. After 48 h of incubation, the cells that had invaded into the lower chamber were collected, stained with 0.1% crystal violet, and quantified. For each insert, five nonoverlapping fields were imaged at 20× magnification (scale bar: 400 μm). Invaded cells were counted via Image J using the “Analyze Particles” function after intensity thresholding, and counts were averaged per insert. Data were obtained from three independent experiments (n = 3).
Real-time quantitative polymerase chain reaction (RT–qPCR)
Total RNA was extracted using TRIzol according to the manufacturer’s protocol. cDNA was synthesized using the PrimeScript RT reagent kit (RR820A, TaKaRa, Dalian, China) in 20-µL reactions containing 2 µg of total RNA, 5× buffer (4 µL), PrimeScript RT Enzyme Mix I (1 µL), Oligo(dT) (1 µL), Random 6-mers (4 µL), and RNase-free water to volume (37°C for 15 min; 85°C for 5 s). Quantitative PCR (qPCR) was conducted using TB Green/SYBR Premix Ex Taq II (2×) in 20-µL reactions comprising premix (10 µL), forward/reverse primers (0.4 µM each; 0.8 µL of 10 µM per primer), 2 µL cDNA (≤10% of total volume), Rhodamine X (ROX; 50×, 0.4 µL) when required, and RNase-free water up to 20 µL. Cycling conditions were 95°C for 30 s, followed by 40 cycles of 95°C for 5 s and 60°C for 34 s; melt-curve analysis confirmed single-product amplification. Primer sequences are listed in Table A2. Relative expression was calculated using the 2−ΔΔCt method with GAPDH as the reference gene. All RT–qPCR measurements were performed in three independent biological replicates (n = 3).
Western blotting (WB)
Cells (2 × 106 per sample) were lysed, and total protein was quantified using the bicinchoninic acid (BCA) assay. For each target, 30 µg of total protein per lane was mixed with 5× loading buffer, denatured at 95°C for 5 min, separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) (10% resolving, 4% stacking), and transferred onto polyvinylidene fluoride (PVDF) membranes (200 mA, 4°C, 1 h). Membranes were blocked with 5% non-fat milk in TBST for 30 min at room temperature, incubated overnight at 4°C with primary antibodies, and then incubated for 1 h at room temperature with horseradish peroxidase (HRP)–conjugated secondary antibodies (Table A3). Bands were visualized using ECL (PK10001; Proteintech, Wuhan, China) and imaged; densitometry was normalized to GAPDH. All blots were repeated in three independent experiments (n = 3).
Chromatin immunoprecipitation (ChIP)
ChIP was performed using a magnetic ChIP kit (26157, Thermo Fisher) according to the manufacturer’s instructions. Cells were lysed and digested, followed by immunoprecipitation with a primary antibody against ATF3 (Table A3). The immunoprecipitate was eluted, and the DNA was collected for RT–qPCR analysis. ChIP–qPCR data were obtained from three independent experiments (n = 3).
Nude mouse model for tumor metastasis evaluation
This study was approved by the Laboratory Animal Ethics Committee of Jiaxing University College of Medicine (899 Guangqiong Road, Jiaxing, Zhejiang, China; Approval No.: JUMC2022-067; 8 April 2022). All procedures complied with Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines, and measures were taken to minimize animal distress. 13
Female nude mice (nu/nu, 5 weeks old; HFK Bioscience, Beijing, China) that were confirmed to be pathogen-free were maintained under specific pathogen-free conditions (12-h light/dark cycle, 22°C ± 2°C, 50%–60% humidity) with ad libitum access to food and water. Animals were acclimatized for 7 days before experimentation.
Mice were randomly assigned to the vector (control) or PERP overexpression group (n = 5 per group) using a computer-generated random number sequence. Cage positions and handling order were randomized to minimize confounding. The sample size was determined based on statistical significance, resource limitations, reproducibility, animal welfare, and findings from a preliminary experiment (4 mice). No animals were excluded. The RAND() function in Excel software was utilized to generate random numbers between 0 and 1. Both groups were housed under identical conditions, and experimenters and data analysts were blinded to group allocation to minimize observation and analysis bias.
Each mouse received an intraperitoneal injection of 6 × 107 cells near the inguinal region. Mice were then maintained for 4 weeks to allow tumor formation. For in vivo imaging, D-luciferin (50 mg/mL, 60 µL) was administered intraperitoneally, after which the animals were anesthetized using gas anesthesia. Following a 10-min incubation, bioluminescent imaging was performed to evaluate tumor growth and metastatic spread.
Patient tissue collection and ethics
This study was approved by the Medical Ethics Committee of The First Hospital of Jiaxing (1882 Zhonghuan South Road, Jiaxing, Zhejiang, China; Approval No.: LS2021-KY-047, 17 May 2021), and written informed consent was obtained from all patients. All procedures involving human participants were conducted in accordance with the ethical standards of the institutional research committee and the Declaration of Helsinki (1975, as revised in 2024). The inclusion criteria were as follows: (a) a diagnosis of primary breast cancer; (b) hospitalization between 1 January 2012 and 31 December 2013; and (c) a histological type confirmed as invasive nonspecial type breast cancer. The exclusion criteria were as follows: (a) age <18 years and (b) prior neoadjuvant chemotherapy. Tumor samples were fixed in 10% neutral buffered formalin for 6–48 h, followed by routine processing steps of dehydration, clearing, wax impregnation, embedding, and storage at room temperature. Patients were monitored, and the primary endpoint was D-DFS. 14
Immunohistochemistry (IHC)
For antigen retrieval, formalin-fixed, paraffin-embedded tumor tissue was dewaxed, hydrated, and heated in citrate buffer for 20 min. To prevent nonspecific binding, 20% goat serum was applied, followed by incubation with 50 µL of anti-PERP primary antibody (HPA022269, MERCK) at 4°C overnight. EnVision reagent and 3,3′-diaminobenzidine (DAB) were used for chromogenic detection, and hematoxylin was used for nuclear counterstaining. Staining intensity (0: none, 1: weak, 2: moderate, and 3: strong) and the proportion of positive cells (0: none, 1: ≤25%, 2: 26%–50%, 3: 51%–75%, and 4: 76%–100%) were scored across five randomly selected fields at 200× magnification (scale bar: 200 μm). 15 Two pathologists independently scored all samples, and discrepancies were resolved by reexamination. The final PERP expression score was calculated as the sum of the intensity and proportion scores.
Statistical analysis
Statistical analyses were performed using SPSS 22.0 (IBM Corp., Armonk, NY, USA), and figures were generated via GraphPad Prism 8.0 (GraphPad Software, San Diego, CA, USA). Data were presented as mean ± SD unless otherwise indicated. Normality was assessed using the Shapiro–Wilk test and homogeneity of variances using Levene’s test prior to applying parametric tests. For comparisons between two groups, Student’s t-test was used for normally distributed data, and the Mann–Whitney U test for non-normal data. Categorical variables were analyzed using Pearson’s χ2 test. Kaplan–Meier survival curves were compared using the log-rank test. All p-values were two-sided, with p < 0.05 considered statistically significant. For bioinformatics analyses, microarray differential expression (GSE45827) was performed in R/limma with Benjamini–Hochberg FDR adjustment, and RNA-seq differential expression was performed in R/DESeq2; significance was defined as FDR < 0.05 and |log2FC| ≥ 1.
Results
PERP is expressed at low levels in breast cancer and is associated with reduced D-DFS
Among 142 tumors, PERP IHC scores ranged from 2 to 7, resulting in 72 (50.7%) PERP-low (scores 2–4) and 70 (49.3%) PERP-high (scores 5–7) cases (Figure 1(a)). The distribution of PERP categories differed significantly across molecular subtypes (χ2 = 16.176, df = 3, p = 0.001), with a higher proportion of PERP-high cases in Luminal A tumors and enrichment of PERP-low cases in TNBC and HER2-positive tumors (Figure 1(b); Table 1). Patients with PERP-low tumors exhibited worse D-DFS than those with PERP-high tumors (log-rank p = 0.029; Figure 1(c)). Consistently, analysis of the GSE45827 dataset revealed that breast cancer subtypes exhibit lower PERP mRNA levels than normal breast tissues (Figure 1(d)). In cell models, PERP expression was reduced in breast cancer lines (MCF-7, MDA-MB-231, and SK-BR-3) relative to MCF-10A, as determined by RT–qPCR and WB (Figure 1(e) and (f)).
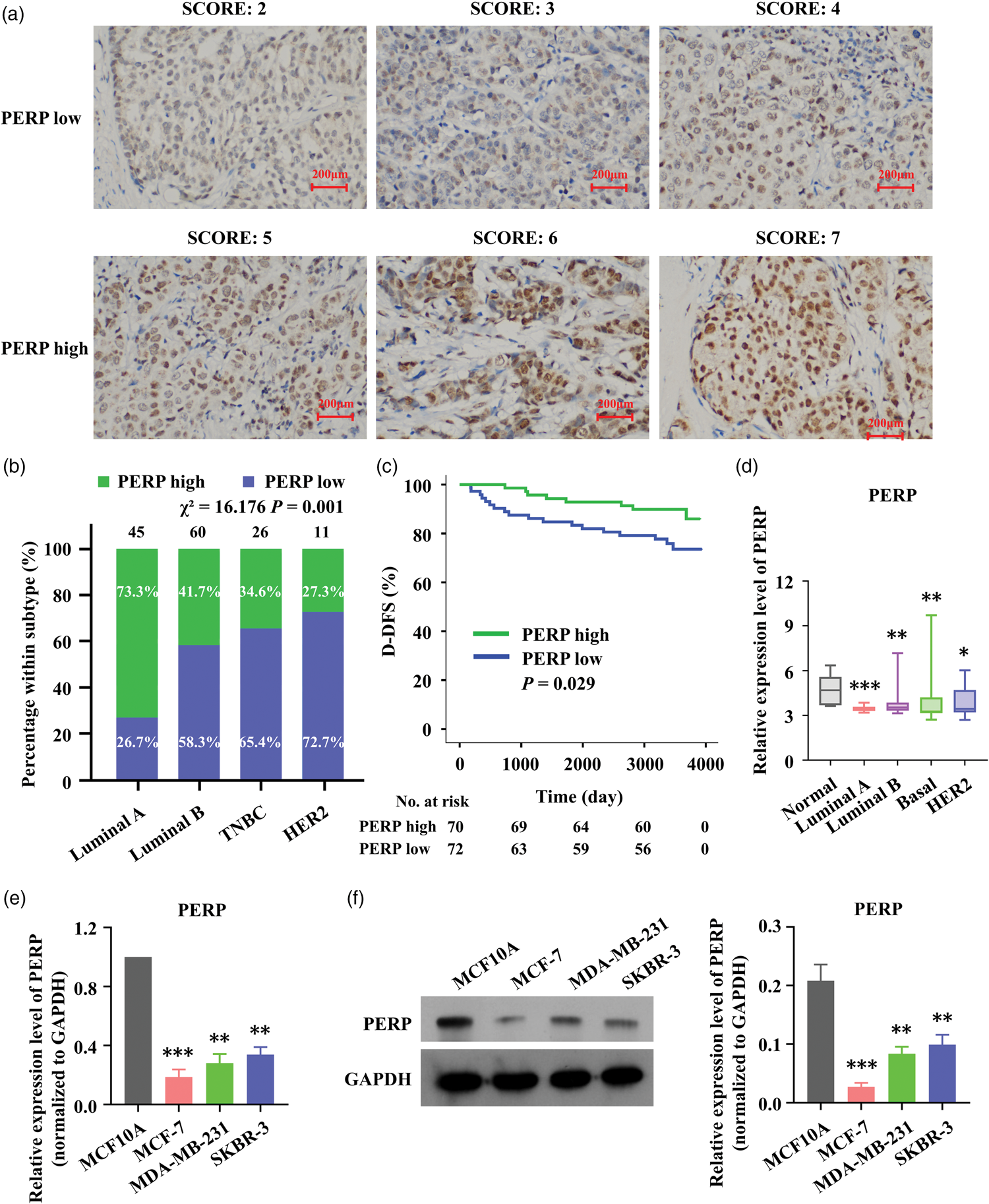
PERP expression is low in breast cancer and is associated with reduced D-DFS. (a) Representative IHC micrographs showing PERP scoring (low: 2–4; high: 5–7); scale bars, 200 μm (200×). (b) Distribution of PERP categories across molecular subtypes (100% stacked). Y-axis indicates percentage within each subtype (%). Labels indicate sample size per subtype and the proportion of PERP-high (green) and PERP-low (blue) tumors. Pearson’s χ2 = 16.176, df = 3, p = 0.001. (c) Kaplan–Meier curves for D-DFS comparing PERP-low versus PERP-high tumors (log-rank p = 0.029). (d) PERP mRNA levels in GSE45827 (normal vs. breast cancer subtypes); boxplots show median and interquartile range and (e–f) PERP expression in cell lines relative to MCF-10A, as measured by RT–qPCR and WB; data represent mean ± SD of three independent experiments. Statistical tests are specified in Methods. *p < 0.05, **p < 0.01, ***p < 0.001. PERP: p53 apoptosis effector related to PMP-22; D-DFS: distant disease-free survival; IHC: immunohistochemistry; RT–qPCR, real-time quantitative polymerase chain reaction; WB: western blotting; SD: standard deviation.
Overexpression of PERP inhibits migration and invasion of breast cancer cells in vitro
We transfected MCF-7 and MDA-MB-231 cells with a PERP overexpression plasmid (oe-PERP) and used an empty plasmid (vector) as a control. PERP expression was evaluated 48 h later using RT–qPCR and WB. The results showed a significant increase in both PERP mRNA and protein levels (Figure 2(a) and (b)), confirming the plasmid’s effectiveness and adequate transfection efficiency. Scratch and Transwell assays revealed that PERP overexpression inhibited cell migration and invasion (Figure 2(c) and (d)).

PERP overexpression suppresses migration and invasion of breast cancer cells in vitro. (a) RT–qPCR analysis showing that transfection with the PERP overexpression plasmid upregulates PERP expression (mean ± SD, n = 3). (b) WB analysis demonstrating increased PERP protein expression following plasmid transfection (densitometry normalized to GAPDH, mean ± SD, n = 3). (c) Scratch assay results showing that PERP overexpression inhibits MCF-7 and MDA-MB-231 cell migration. Phase-contrast images were acquired with a 10× objective; scale bar, 200 µm; three pre-marked fields per well were analyzed and (d) transwell invasion assay showing reduced invasion upon PERP overexpression in MCF-7 and MDA-MB-231 cells. Cells were stained with 0.1% crystal violet, images were acquired at 20×; scale bar, 400 µm; five nonoverlapping fields per insert were quantified using ImageJ (mean ± SD, n = 3). *p < 0.05, **p < 0.01, ***p < 0.001. PERP: p53 apoptosis effector related to PMP-22; RT–qPCR: real-time quantitative polymerase chain reaction; WB: western blotting; GAPDH: glyceraldehyde-3-phosphate dehydrogenase.
Overexpression of PERP promotes HSPA6 expression
To explore PERP-regulated pathways, we performed RNA-seq for transcriptomic analysis. MDA-MB-231 cells transfected with the PERP overexpression plasmid served as the experimental group, and those transfected with an empty plasmid were used as a control. Compared with the control, 3769 genes were upregulated and 1555 genes were downregulated in the PERP group, with HSPA6 showing the greatest upregulation (Figure 3(a)). WB confirmed that PERP overexpression significantly upregulated HSPA6 expression in both MCF-7 and MDA-MB-231 cells, consistent with the RNA-seq results (Figure 3(b)).

PERP overexpression promotes HSPA6 expression. (a) Scatter plot comparing gene-wise mean expression (log2 scale) between conditions (x-axis: vector mean; y-axis: PERP mean). The diagonal represents equal expression. Points are colored according to regulation status from differential expression analysis. PERP, ATF3, and HSPA6 are highlighted with labeled markers, illustrating strong upregulation of the PERP–ATF3–HSPA6 axis and (b) WB analysis showing that transfection with the PERP overexpression plasmid upregulates HSPA6 protein expression (densitometry normalized to GAPDH, mean ± SD, n = 3). ***p < 0.001. PERP: p53 apoptosis effector related to PMP-22; HSPA6: heat shock protein family A (Hsp70) member 6; ATF3: activating transcription factor 3; WB: western blotting; GAPDH: glyceraldehyde-3-phosphate dehydrogenase.
PERP enhances HSPA6 expression via ATF3
Using the hTFtarget database, we predicted that 106 transcription factors potentially bind to the HSPA6 promoter. By integrating these predictions with RNA-seq data, we identified 10 candidate transcription factors, with ATF3 exhibiting the most significant upregulation upon PERP overexpression (Figure 4(a)).

PERP enhances HSPA6 expression via ATF3. (a) ATF3 was identified as a potential transcription factor regulating HSPA6 expression based on RNA-seq analysis and transcription factor prediction. (b) WB analysis showing that transfection with the PERP overexpression plasmid upregulates ATF3 protein expression (densitometry normalized to GAPDH, mean ± SD, n = 3). (c) RT–qPCR showing that ATF3 Continued.knockdown suppresses HSPA6 mRNA expression (mean ± SD, n = 3). (d) WB analysis demonstrating that ATF3 knockdown reduces HSPA6 expression (densitometry normalized to GAPDH, mean ± SD, n = 3). (e) ChIP assay showing that the P1 region exhibits the highest affinity for ATF3 binding (mean ± SD, n = 3). (f) RT–qPCR indicating that ATF3 downregulation prevents the PERP-mediated increase in HSPA6 mRNA expression (mean ± SD, n = 3) and (g) WB indicating that ATF3 downregulation blocks the PERP-induced increase in HSPA6 protein expression (densitometry normalized to GAPDH, mean ± SD, n = 3). *p < 0.05, **p < 0.01, ***p < 0.001. PERP: p53 apoptosis effector related to PMP-22; HSPA6: heat shock protein family A (Hsp70) member 6; ATF3: activating transcription factor 3; RNA-seq: RNA sequencing; WB: western blotting; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; RT–qPCR: real-time quantitative polymerase chain reaction; ChIP: chromatin immunoprecipitation.
PERP overexpression in MDA-MB-231 cells increased ATF3 expression, as confirmed by WB (Figure 4(b)). siRNA-mediated knockdown of ATF3 (si-ATF3) reduced ATF3 expression, as validated by RT–qPCR and WB, and correspondingly decreased HSPA6 levels (Figure 4(c) and (d)). ATF3 depletion did not affect PERP protein abundance in MDA-MB-231 cells (si-ATF3 vs. si-NC), as shown by immunoblotting (Figure A1). These findings support a model in which PERP upregulates HSPA6 via ATF3, while ATF3 does not respond to regulate PERP levels.
JASPAR database analysis identified three potential ATF3 binding sites in the HSPA6 promoter (Table A4). ChIP analysis revealed that the P1 region exhibited the highest affinity for ATF3 binding (Figure 4(e)), confirming ATF3 as a transcription factor for HSPA6. Furthermore, ATF3 knockdown partially reversed PERP-mediated HSPA6 upregulation in MCF-7 and MDA-MB-231 cells (Figure 4(f) and (g)), indicating that PERP-induced HSPA6 upregulation is mediated via ATF3.
PERP inhibits breast cancer cell migration and invasion in vitro through the ATF3/HSPA6 signaling axis
An siRNA targeting HSPA6 (si-HSPA6) was constructed and transfected into MDA-MB-231 cells, using si-NC as a control. RT–qPCR and WB showed that si-HSPA6 effectively reduced HSPA6 expression (Figure 5(a) and (b)), validating both the specificity and transfection efficiency of si-HSPA6. The vectors oe-PERP, si-NC, si-ATF3, and si-HSPA6 were transfected into MCF-7 and MDA-MB-231 cells. Scratch and Transwell assays demonstrated that knockdown of ATF3 or HSPA6 partially reversed PERP’s inhibitory effects on cell migration and invasion, indicating that PERP suppresses breast cancer cell migration and invasion via the ATF3/HSPA6 axis (Figure 5(c) and (d)).

PERP inhibits breast cancer cell migration and invasion in vitro via the ATF3/HSPA6 signaling axis. (a) RT–qPCR showing that si-HSPA6 transfection reduces HSPA6 expression (mean ± SD, n = 3). (b) WB analysis demonstrating that si-HSPA6 transfection decreases HSPA6 protein levels (densitometry normalized to GAPDH, mean ± SD, n = 3). (c) Scratch assay showing that downregulation of ATF3 or HSPA6 partially reverses the PERP-mediated inhibition of cell migration (phase-contrast images acquired with a 10× Continued.objective; scale bar, 200 µm; three pre-marked fields per well) and (d) transwell assay demonstrating that ATF3 or HSPA6 knockdown attenuates PERP-mediated suppression of cell invasion (stained with 0.1% crystal violet; images at 20× magnification; scale bar, 400 µm; five nonoverlapping fields per insert, cell counts via ImageJ, mean ± SD, n = 3). *p < 0.05, **p < 0.01, ***p < 0.001. PERP: p53 apoptosis effector related to PMP-22 ATF3: activating transcription factor 3; HSPA6: heat shock protein family A (Hsp70) member 6; RT–qPCR: real-time quantitative polymerase chain reaction; WB: western blotting; GAPDH: glyceraldehyde-3-phosphate dehydrogenase.
PERP reduces intraperitoneal tumor burden in vivo
We transfected PERP-overexpressing lentivirus into MDA-MB-231 cells, with successful transfection confirmed via fluorescence microscopy (Figure 6(a)) and PERP overexpression validated using RT–qPCR (Figure 6(b)) and WB (Figure 6(c)). In an intraperitoneal dissemination model, mice injected intraperitoneally with oe-PERP cells exhibited lower bioluminescent signals than controls under identical acquisition settings (Figure 6(d); n = 5 per group), indicating a reduced overall intraperitoneal tumor burden.
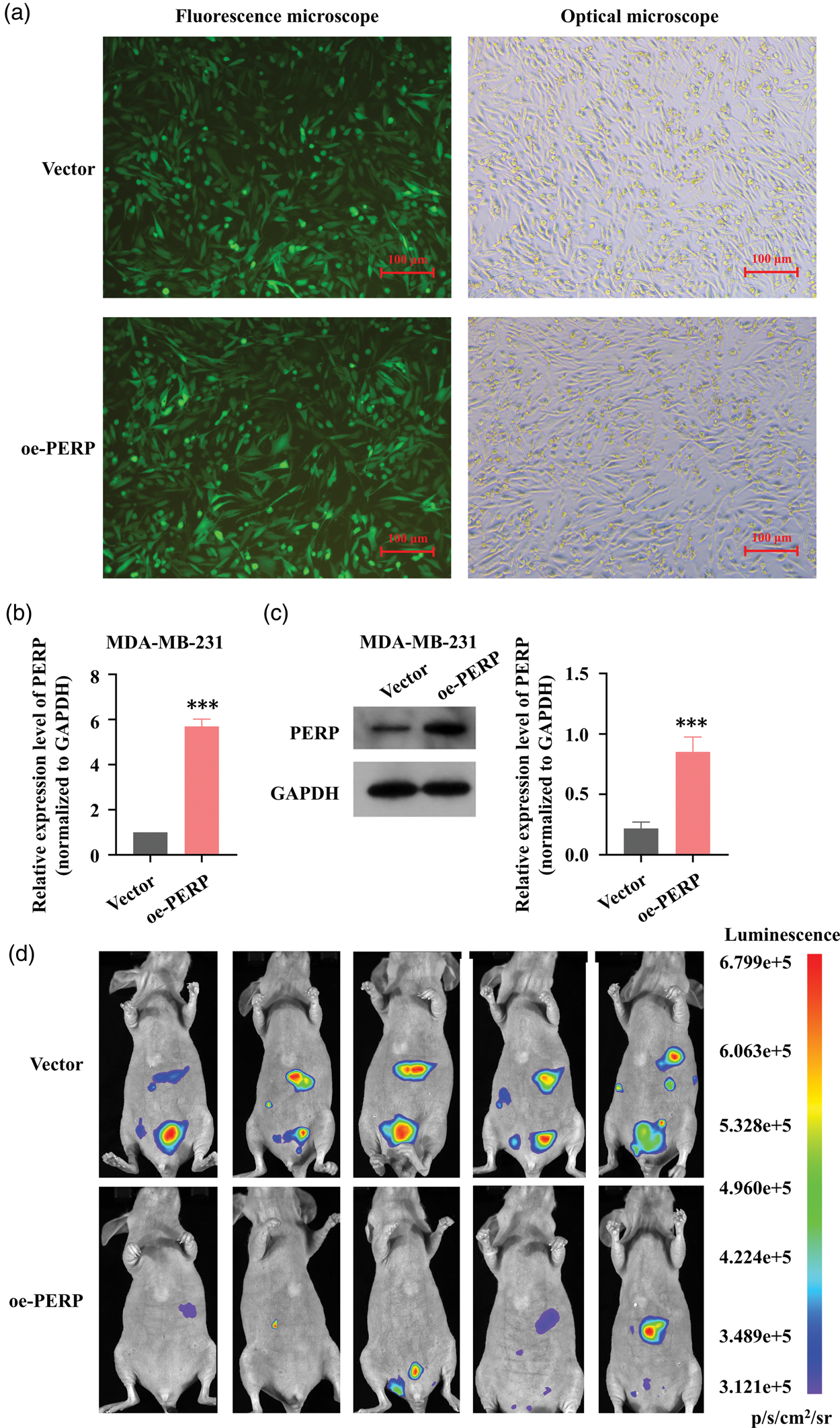
PERP reduces intraperitoneal tumor burden in vivo. (a) Fluorescence microscopy confirmed successful transduction of fluorescent lentiviral particles into breast cancer cells (color scale indicates photons/s; 20× magnification; scale bar, 100 µm). (b) RT–qPCR analysis showing that MDA-MB-231 cells Continued.transduced with oe-PERP lentivirus exhibited elevated PERP expression (mean ± SD, n = 3). (c) WB analysis demonstrating that oe-PERP lentivirus increased PERP expression in MDA-MB-231 cells (densitometry normalized to GAPDH, mean ± SD, n = 3) and (d) in vivo intraperitoneal dissemination model showing reduced tumor burden in mice injected with PERP-overexpressing cells (n = 5). ***p < 0.001. PERP: p53 apoptosis effector related to PMP-22; RT–qPCR: real-time quantitative polymerase chain reaction; WB: western blotting; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; oe: overexpression.
Discussion
PERP is a p53-regulated membrane protein localized to desmosomes, where it maintains epithelial cohesion and tissue integrity.16,17 Across different malignancies, PERP can act as a tumor suppressor or, in a context-dependent manner, be associated with poor outcomes, highlighting pathway-specific effects and biological heterogeneity.4,5,18–21 Despite this complexity, the impact of PERP dysregulation on metastatic behavior has been less extensively studied than its roles in proliferation and apoptosis.
We delineate a PERP→ATF3→HSPA6 regulatory axis that limits breast cancer cell motility. Mechanistically, PERP upregulates the expression of the stress-responsive transcription factor ATF3, which binds to the HSPA6 promoter and enhances HSPA6 expression; functionally, knockdown of ATF3 or HSPA6 attenuates PERP-mediated antimigratory/anti-invasive effects. This pathway links desmosomal/adhesion cues (PERP) to a stress-responsive chaperone program (ATF3/HSPA6) and provides a mechanistic route by which epithelial integrity can constrain cellular motility.
Our findings align with previous observations. Consistent with Dusek et al., who reported reduced PERP expression in breast cancer relative to nonmalignant epithelium, 22 PERP deficiency provides a plausible entry point for motility-promoting programs. Furthermore, studies demonstrating that diminished PERP weakens cell–cell adhesion and facilitates EMT, thereby enhancing invasion and metastasis, 23 complement our axis by positioning PERP at the interface of adhesion homeostasis and migratory regulation. Consistent with the model-specific roles reported for HSPA6 across tumor types,24–28 our data indicate that HSPA6 acts as a motility-restraining effector in this context. Within this framework, the apparently dualistic behavior of ATF3 in breast cancer may be interpreted as context-dependent target selection.29–32
From a translational perspective, the association between low PERP expression and adverse outcomes highlights opportunities for biomarker development and restorative strategies. Interventions that stabilize or upregulate PERP or those that mimic its downstream program (e.g. ATF3/HSPA6 activation) could be investigated as metastasis-targeted therapies while considering cancer-type specificity and the need for patient stratification. This axis also suggests pharmacodynamic readouts, including ATF3/HSPA6 levels and quantitative motility metrics, for early-phase studies.
Although previous studies link HSPA6 to PI3K–AKT and NF-κB signaling, our transcriptomic dataset under the present conditions did not reveal a strong, concordant activation signature. Therefore, the mechanistic analysis was based on the PERP→ATF3→HSPA6 cascade. Targeted assays—such as phospho-AKT immunoblotting, NF-κB p65 nuclear translocation/reporter analyses, and HSPA6 gain- or loss-of-function rescue experiments—are warranted to evaluate whether these pathways contribute to HSPA6-dependent effects in additional contexts.
Although HSPA6 has been associated with proliferation and survival in certain models, our focus here was to delineate the PERP→ATF3→HSPA6 axis in the context of cell motility. Because PERP’s antiproliferative function has been previously established—including our own work identifying DCAF13 as an upstream regulator that promotes proliferation via PERP suppression—we did not re-evaluate growth/survival endpoints in this study. To avoid redundancy, we prioritized nonoverlapping migration/invasion readouts; nevertheless, proliferation/survival effects are likely context-dependent and warrant dedicated assessment under conditions such as stress, chemotherapy exposure, extended time frames, and additional models.
This study has several limitations. To better harmonize expression systems, repeating key in vitro assays with PERP-stable (or inducible) lines would be informative; this was not undertaken in the present study due to resource constraints but is planned for future work. In vivo, we employed a single breast cancer line and an ethically approved intraperitoneal model; subsequent studies should incorporate orthotopic or tail-vein models with endpoint histological confirmation to assess organ tropism and validate metastatic foci.
In summary, this study identifies PERP as a suppressor of metastatic traits in breast cancer via ATF3-dependent induction of HSPA6, delineating an adhesion-to-stress signaling pathway that limits cell motility (Figure 7). This framework highlights a potential tractable axis for biomarker development and therapeutic modulation.

Schematic model illustrating PERP-mediated suppression of breast cancer metastasis. PERP upregulates the expression of ATF3, which binds to the HSPA6 promoter and enhances HSPA6 transcription, ultimately inhibiting breast cancer cell migration and invasion. PERP: p53 apoptosis effector related to PMP-22; ATF3: activating transcription factor 3; HSPA6: heat shock protein family A (Hsp70) member 6.
Supplemental Material
sj-pdf-1-imr-10.1177_03000605251406800 - Supplemental material for PERP suppresses breast cancer metastasis via the ATF3–HSPA6 signaling pathway
Supplemental material, sj-pdf-1-imr-10.1177_03000605251406800 for PERP suppresses breast cancer metastasis via the ATF3–HSPA6 signaling pathway by Caiping Chen, Chao Han, Li Xue, Wanxin Wu, Lin Zhang and Xiang Lu in Journal of International Medical Research
Supplemental Material
sj-pdf-2-imr-10.1177_03000605251406800 - Supplemental material for PERP suppresses breast cancer metastasis via the ATF3–HSPA6 signaling pathway
Supplemental material, sj-pdf-2-imr-10.1177_03000605251406800 for PERP suppresses breast cancer metastasis via the ATF3–HSPA6 signaling pathway by Caiping Chen, Chao Han, Li Xue, Wanxin Wu, Lin Zhang and Xiang Lu in Journal of International Medical Research
Supplemental Material
sj-pdf-3-imr-10.1177_03000605251406800 - Supplemental material for PERP suppresses breast cancer metastasis via the ATF3–HSPA6 signaling pathway
Supplemental material, sj-pdf-3-imr-10.1177_03000605251406800 for PERP suppresses breast cancer metastasis via the ATF3–HSPA6 signaling pathway by Caiping Chen, Chao Han, Li Xue, Wanxin Wu, Lin Zhang and Xiang Lu in Journal of International Medical Research
Supplemental Material
sj-pdf-4-imr-10.1177_03000605251406800 - Supplemental material for PERP suppresses breast cancer metastasis via the ATF3–HSPA6 signaling pathway
Supplemental material, sj-pdf-4-imr-10.1177_03000605251406800 for PERP suppresses breast cancer metastasis via the ATF3–HSPA6 signaling pathway by Caiping Chen, Chao Han, Li Xue, Wanxin Wu, Lin Zhang and Xiang Lu in Journal of International Medical Research
Supplemental Material
sj-pdf-5-imr-10.1177_03000605251406800 - Supplemental material for PERP suppresses breast cancer metastasis via the ATF3–HSPA6 signaling pathway
Supplemental material, sj-pdf-5-imr-10.1177_03000605251406800 for PERP suppresses breast cancer metastasis via the ATF3–HSPA6 signaling pathway by Caiping Chen, Chao Han, Li Xue, Wanxin Wu, Lin Zhang and Xiang Lu in Journal of International Medical Research
Footnotes
Acknowledgments
Not applicable.
Author contributions
All authors contributed to the study conception and design. Data curation was performed by Caiping Chen, Li Xue, Chao Han, and Wanxin Wu. Funding acquisition was performed by Caiping Chen and Xiang Lu. Writing–original draft was performed by Caiping Chen. Methodology was performed by Li Xue. Investigation was performed by Chao Han and Lin Zhang. Validation was performed by Xiang Lu. Formal analysis was performed by Wanxin Wu and Xiang Lu. Visualization was performed by Wanxin Wu. Conceptualization, project administration, resources, software, supervision, and writing–review & editing were performed by Xiang Lu. All authors reviewed the manuscript and approved the final submitted version.
Consent to participate and consent to publish
Written informed consent was obtained from the volunteer and their family members prior to using the data in this study.
Data availability statement
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Declaration of conflicting interests
The authors declare no conflicts of interest.
Ethics statement
The research protocol was approved by the Ethics Committee of The First Hospital of Jiaxing (Approval No.: LS2021-KY-047, 17 May 2021).
Written informed consent was obtained from all patients.
Animal experiments were approved by the Ethics Committee of Jiaxing University (Approval No.: JUMC2022-067, 8 April 2022).
Funding
This work was supported by the Jiaxing Science and Technology Plan Project (2022AY30019); Research Fund of the First Hospital of Jiaxing (2023-YB-036); Jiaxing Key Discipline of Medicine (Mastropathy, Innovation Subject) (2023-FC-001); Breast Cancer Precision Diagnosis and Treatment Center of the First Hospital of Jiaxing (2021-ZZZX-06); Jiaxing Provinces and Cities Jointly Cultivate Discipline–General Surgery (Minimally Invasive) (2023-PYXK-001); Jiaxing Key Discipline of Traditional Chinese Medicine Surgery of Traditional Chinese Medicine (2023-ZYYCX-002); and Clinical Specialist Professional Skills Innovation and Application Research Project (RCLX2315084).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.