
Abstract
Background
This study employed Mendelian randomization to investigate the relationships between pathogenic infections, immunophenotypes, and Hashimoto’s thyroiditis, providing deeper insights into infection-induced Hashimoto’s thyroiditis development beyond the limitations of small, inconclusive observational studies.
Methods
Data on pathogenic infections, immune cells, and Hashimoto’s thyroiditis were obtained from public databases. The inverse variance weighted method was used as the primary analytical approach, with robustness of the findings confirmed through heterogeneity and pleiotropy tests.
Results
Mendelian randomization analysis demonstrated a causal relationship between anti-polyomavirus 2 IgG seropositivity and Hashimoto’s thyroiditis (inverse variance weighted: odds ratio = 1.145, 95% confidence interval: 1.069–1.225, p = 9.90e−05). There was insufficient evidence to support a reverse causal relationship (inverse variance weighted: odds ratio = 1.092, 95% confidence interval: 0.892–1.337, p = 3.94e−01). The proportion of variation in genetically predicted anti-polyomavirus 2 IgG seropositivity mediated by CD20+ IgD+ CD38− B cells was 6.36% (95% confidence interval: 1.38%–11.35%).
Conclusion
Mendelian randomization analysis revealed that polyomavirus 2 infection significantly contributed to the development of Hashimoto’s thyroiditis, mediated by CD20+ IgD+ CD38⁻ B cells. However, no causal associations were observed between Hashimoto’s thyroiditis and other commonly studied pathogens, including human herpesvirus 6, hepatitis C virus, Epstein–Barr virus, and Helicobacter pylori.
Keywords
Introduction
Hashimoto’s thyroiditis (HT), also known as chronic lymphocytic thyroiditis, affects approximately 10%–12% of the global population.1,2 The exact causes of HT remain incompletely understood; however, they are known to involve genetic, environmental, and epigenetic factors. 3 Due to its unclear pathogenesis, strategies to prevent thyroid tissue damage and dysfunction in affected patients are currently limited. 4 Understanding the etiology of HT is essential for developing effective preventive and therapeutic approaches.
Pathogen infections have been associated with a wide range of autoimmune diseases.5,6 Among these, HT, one of the most prevalent autoimmune disorders, has garnered increasing attention regarding the role of pathogen infections in its pathogenesis. Specifically, the involvement of human herpesvirus (HHV) 6,7,8 Epstein–Barr virus,9–11 Helicobacter pylori infection,12–14 and hepatitis C virus15–18 in the development of HT has attracted significant attention. The prevailing hypothesis suggests that pathogens trigger inflammatory cellular immune responses through molecular mimicry, thereby influencing the role of immune cells in the onset of HT.19,20 However, most of these studies were conducted on relatively small patient cohorts and lacked internal controls, a limitation often resulting in inconsistent results. 21 For instance, Figura et al. reported that H. pylori infection is a risk factor for HT, 12 whereas Shmuely et al. did not replicate these findings. 14 Consequently, a direct link between viral etiology and the onset of HT has not been definitively established,3,15,19–21 as attempts to detect viruses within the thyroid tissue of patients with HT have failed to yield reliable results.3,21
Concurrently, an increasing number of studies are elucidating the complex interactions between the immune system and HT. 22 According to the molecular mimicry hypothesis, immune cells may play a critical role in mediating the pathways that link pathogen infections to HT. Given the inherent limitations of observational studies, including unmeasured or inaccurately measured confounding factors, reverse causality, and various sources of bias, 23 there is an urgent need for innovative research methodologies to investigate the interactions between pathogen infections, immune cells, and HT. Such approaches could provide a robust foundation for the diagnosis and treatment of HT.
Mendelian randomization (MR) is an advanced approach that employs genetic variants as instrumental variables (IVs) to infer causal relationships from observational data, thereby accurately assessing the impact of exposure factors on outcomes. 24 This strategy draws on the principle that genetic variants are randomly distributed at conception, creating a framework similar to a natural experiment. MR analysis enables researchers to explore potential causal links between risk factors and disease outcomes, whereas the random distribution of genetics helps mitigate the influence of confounding factors. 25 This approach effectively addresses challenges posed by unmeasured confounders or biases, thereby accurately circumventing issues of reverse causality. 26
In our study, we utilized the latest summary statistics from genome-wide association studies (GWAS) to investigate the causal relationships between various pathogen infections and HT. Through mediator MR analysis, we examined the pivotal role of immunophenotypes in this process. To the best of our knowledge, this is the first study to employ MR analysis to investigate the relationships between multiple pathogens and HT. Our findings enhance the understanding of the pathogenesis of HT and provides a scientific foundation for the diagnosis and treatment of HT.
Method
Study design
The aim of this study was to explore the causal relationship between various pathogens and HT as well as to investigate the mediating role of immune cells in this process, using a two-sample bidirectional MR analysis. All research procedures were conducted in strict accordance with the Strengthening the Reporting of Observational Studies in Epidemiology using Mendelian Randomization (STROBE-MR) guidelines. 27 In this study, we used single nucleotide polymorphisms (SNPs) as IVs for MR analysis. 26 The selection of SNPs must meet three criteria: (a) a strong association with the exposure of interest; (b) no association with any potential confounders of the exposure and outcome; and (c) no direct association with the outcome other than through the exposure. 28
As all data used in this study were publicly available and had received approval from the relevant ethics review committee, no additional ethical approval was required.
Data source
Data for pathogen infection
In this study, we acquired data on pathogen infections by measuring immune responses and antibody-mediated reactions following infection (Table 1). The GWAS data were primarily sourced from the UK Biobank, encompassing serological measurements for up to 10,000 infectious disease cases alongside whole-genome genotyping data. Butler-Laporte et al. defined 46 disease phenotypes based on data from 13 pathogens, including 15 seropositive case–control phenotypes and 31 quantitative antibody measurement phenotypes. 29 Particularly, these 13 pathogens included herpes simplex virus type 1, herpes simplex virus type 2, Epstein–Barr Virus, human cytomegalovirus, HHV-6, HHV-7, Varicella zoster virus, human BK polyomavirus, human JC polyomavirus, Merkel cell polyomavirus, Chlamydia trachomatis, H. pylori, and Toxoplasma gondii. Additional details are provided in Supplementary Table S1.
Details of GWAS and datasets used in the analyses.

IAMIR: infections and antibody-mediated immune responses; GWAS: genome-wide association.
Through literature review, we identified hepatitis C infection as a potential contributor to the development of HT. Due to the absence of hepatitis C data in the primary dataset, we sourced summary data on hepatitis C infections from two East Asian cohorts available at https://gwas.mrcieu.ac.uk. The dataset by Sakaue et al. (ID: ebi-a-GCST90018585) included data from 7110 patients with hepatitis C and 169,588 controls, whereas the dataset by Ishigaki et al. (GWAS ID: bbj-a-101) comprised 5794 patients with hepatitis C and 206,659 controls (Table 1).
Data for HT
Data on HT were obtained from the GWAS summary data compiled by Sakaue et al., which included 15,654 HT cases from European populations and 379,986 European control samples. 30 This study integrated datasets from multiple sources, particularly including significant data from BioBank Japan. To further expand the scope of the analysis, the research team also collaborated with the UK Biobank and the FinnGen project to perform a meta-analysis (Table 1).
Data for 731 immunophenotypes
Immune trait data were obtained from the GWAS Catalog (GCST90001391–GCST90002121), encompassing non-overlapping data from 3757 European individuals. 31 Approximately 22 million SNPs were imputed and analyzed using a Sardinian sequence reference panel. 32 A total of 731 immune phenotypes were examined, including absolute cell (AC) counts (n = 118), median fluorescence intensities (MFI) representing surface antigen levels (n = 389), morphological parameters ((MPs) n = 32), and relative cell (RC) counts (n = 192). The MFI, AC, and RC features included B cells, CDCs, mature T-cell stages, monocytes, myeloid cells, TBNK (T cells, B cells, and natural killer cells), and Treg panels, whereas the MP feature comprised CDC and TBNK panels. Detailed information on 731 immunophenotypes is provided in Supplementary Table S2.
IV selection and data harmonization
When selecting SNPs strongly associated with disease as IVs for MR analysis, we prioritized those with a genome-wide significance threshold of p < 5e−8. However, when the number of SNPs was insufficient for analysis, we appropriately lowered the threshold to p < 5e−6 to include more SNPs. 33
Subsequently, we grouped SNPs based on linkage disequilibrium, using a window size of 10,000 kb and an r2 threshold of <0.001. Moreover, we calculated the F-statistic using the following formula to assess the proportion of variance explained by each candidate SNP.

Finally, based on literature review, we evaluated all known phenotypes associated with the selected genetic instruments and manually excluded SNPs that were potentially linked to confounding factors, including smoking, vitamin D deficiency, postpartum depression, and the use of calcium channel blockers and diuretics.36–38
Primary analysis
Figure 1 provides a schematic overview of this study. Initially, we conducted a two-sample bidirectional MR analysis to assess the mutual causal relationships between various pathogen infections and HT (Figure 1(a)), which we defined as the overall effect.

Diagrams illustrating associations examined in this study. (a) Total effect between pathogen infections and Hashimoto’s thyroiditis (HT). “c” represents the total effect using genetically predicted pathogen infections as the exposure and HT as the outcome. “d” represents the total effect using genetically predicted HT as the exposure and pathogen infections as the outcome. (b) Decomposition of the total effect: (i) indirect effect using a two-step approach (where “a” is the total effect of pathogen infections on immunophenotypes, and “b” is the effect of immunophenotypes on HT) and the product method (a × b); (ii) direct effect (c′ = c − a × b). The proportion mediated is calculated as the indirect effect divided by the total effect.
The inverse variance weighted (IVW) method formed the core of the analytical strategy of this study, combining meta-analysis techniques with the Wald estimates of individual SNPs. Assuming no horizontal pleiotropy, the IVW method can provide unbiased results, with significance set at p value of <0.05.35,39 To enhance the robustness of the findings, additional methods were employed, including Bayesian-weighted Mendelian randomization (BWMR) and the weighted median approach. The BWMR method is particularly suitable for causal inference, effectively managing uncertainties arising from minor effects in polygenic traits. Through Bayesian weighting, it effectively identifies outliers and mitigates the impact of violating of the stringent IV assumptions. 40 Moreover, the weighted median approach offers enhanced precision, evidenced by a reduced standard deviation relative to MR-Egger analysis. Remarkably, even in the presence of horizontal pleiotropy, the weighted median method maintains a consistent estimation, reliable even when up to 50% of the genetic variants are deemed invalid IVs.41,42
Mediation analysis
We employed a two-step MR analysis to investigate whether immunophenotypes mediate the relationship between pathogen infection and HT. The overall effect can be partitioned into indirect effects (a × b in Figure 1(b)) and direct effects (c′ in Figure 1(b)). 43 By calculating the ratio of the indirect effect to the overall effect, we determined the percentage of the overall effect that is mediated. The 95% confidence intervals (CIs) were calculated using the delta method.
Sensitivity analysis
Due to variations in experimental conditions, selected populations, and SNPs, two-sample MR analysis results may exhibit heterogeneity, potentially resulting in biased estimates of causal effects. To address this, heterogeneity was assessed using the IVW and MR-Egger methods. Cochrane’s Q statistic was used to evaluate heterogeneity across genetic instruments, with p value of >0.05 indicating no significant heterogeneity. 44
A key assumption of MR analysis is that IVs influence the outcome solely through exposure, necessitating the assessment of horizontal pleiotropy between exposure and outcome. We employed the MR-Egger intercept method to assess the presence of pleiotropy, where a p value of >0.05 suggests minimal or negligible pleiotropy, allowing for its exclusion from the analysis. 45
Finally, we identified and corrected outliers in the IVW analysis using the MR-PRESSO test 46 and employed “leave-one-out” analysis to evaluate the genetic causal effects of individual SNPs on the exposure–outcome relationship. 47
Statistical analysis
We conducted all the aforementioned MR analyses using R software (version 4.2.0; http://www.r-project.org) and the ‘TwoSampleMR’ package (version 0.6.8).
Results
The study flowchart is presented in Figure 2. The characteristics of significant SNPs used in this study are provided in Supplementary Table S3.

Workflow diagram of the study. IVW: inverse variance weighting; MR: Mendelian randomization; BWMR: Bayesian-weighted Mendelian randomization.
Association of pathogen infection with HT
To investigate the causal relationship between pathogen infection and HT, multiple pathogen infection phenotypes were selected as exposures, with HT as the outcome for MR analysis. Using a genome-wide significance threshold of p < 5e−8 to select SNPs closely associated with the exposures, an insufficient number of IVs was identified. Therefore, based on previous studies, the threshold was adjusted to p < 5e−6.
After screening with the IVW method, no causal relationship was observed between HHV-6, Epstein–Barr virus, and H. pylori infection and HT. Furthermore, incorporating GWAS data from two hepatitis C infection cohorts similarly revealed no causal link between hepatitis C infection and HT (Supplementary Table S4). These findings challenge the conclusions drawn from previous studies.
The results demonstrated that, among the evaluated pathogen infection phenotypes, only anti-polyomavirus 2 IgG seropositivity exhibited a causal relationship with HT (IVW: odds ratio (OR) = 1.145, 95% CI: 1.069–1.225, p = 9.90e−05; weighted median: OR = 1.127, 95% CI: 1.022–1.242, p = 1.64e−02; BWMR: OR = 1.147, 95% CI: 1.067–1.234, p = 1.93e−04) (Figure 3).

Forest plot visualizing the causal effects of CD20+ IgD+ CD38− B cells on anti-polyomavirus 2 IgG seropositivity and HT. HT: Hashimoto’s thyroiditis; IVW: inverse variance weighted; BWMR: Bayesian-weighted Mendelian randomization.
To examine the potential reverse causal relationship between anti-polyomavirus 2 IgG seropositivity and HT, we conducted a reverse MR analysis, considering HT as the exposure and anti-polyomavirus 2 IgG seropositivity as the outcome. To enhance the reliability of our results, we selected SNPs closely associated with the exposure using a genome-wide significance threshold of p < 5e−8 and excluded three outliers: rs3184504, rs7030280, and rs9271365. The results indicated no reverse causal relationship (IVW: OR = 1.092, 95% CI: 0.892–1.337, p = 3.94e−01; weighted median: OR = 1.068, 95% CI: 0.825–1.383, p = 6.16e−01; BWMR: OR = 1.081, 95% CI: 0.922–1.268, p = 3.36e−01) (Figure 3). Detailed results are provided in Supplementary Table S5.
Exploration of mediating factors
Immunophenotypes
To assess whether 731 immunophenotypes act as mediators in the relationship between polyomavirus 2 infection and HT, we initially used anti-polyomavirus 2 IgG seropositivity as the exposure and the 731 immunophenotypes as the outcomes in a MR analysis.
To ensure sufficient number of IVs for the analysis, we applied a genome-wide significance threshold of p < 5e−6 to select SNPs closely associated with the exposure. After filtering using the IVW method, we identified a causal relationship between polyomavirus 2 infection and 23 immunophenotypes (Supplementary Table S6).
Subsequently, we considered these 23 immunophenotypes as the exposure and used a genome-wide significance threshold of p < 5e−6 to select SNPs as IVs, with HT as the outcome, for MR analysis (Supplementary Table S7). Our findings indicated a negative causal relationship between CD20+ IgD+ CD38− B cells and HT (IVW: OR = 0.890, 95% CI: 0.801–0.989, p = 3.06e−02; weighted median: OR = 0.895, 95% CI: 0.775–1.035, p = 1.34e−01; BWMR: OR = 0.887, 95% CI: 0.791–0.996, p = 4.25e−02) (Figure 3).
To investigate whether CD20+ IgD+ CD38− B cells serve as mediators in the pathway from polyomavirus 2 infection to HT, we examined the causal relationship between polyomavirus 2 infection and these cells. Notably, a negative correlation was observed between anti-polyomavirus 2 IgG seropositivity and CD20+ IgD+ CD38− B cells (IVW: OR = 0.929, 95% CI: 0.869–0.992, p = 2.89e−02; weighted median: OR = 0.944, 95% CI: 0.861–1.035, p = 2.21e−01; BWMR: OR = 0.934, 95% CI: 0.872–1.001, p = 5.25e−02) (Figure 3). Therefore, CD20+ IgD+ CD38− B cells can serve as mediators in the pathway from polyomavirus 2 infection to HT.
Proportion of the association between polyomavirus 2 infection and HT mediated by CD20+ IgD+ CD38− B cells
Our analysis indicates that CD20+ IgD+ CD38− B cells can serve as mediators in the pathway from polyomavirus 2 infection to HT. We observed a negative causal relationship between polyomavirus 2 infection and CD20+ IgD+ CD38− B cells as well as between these cells and HT. As shown in Figure 4, the indirect effect was 0.009, accounting for a 6.36% increase in the risk of HT associated with polyomavirus 2 infection (proportion mediated, 6.36%; 95% CI: 1.38%–11.35%), whereas the direct effect was 0.127.
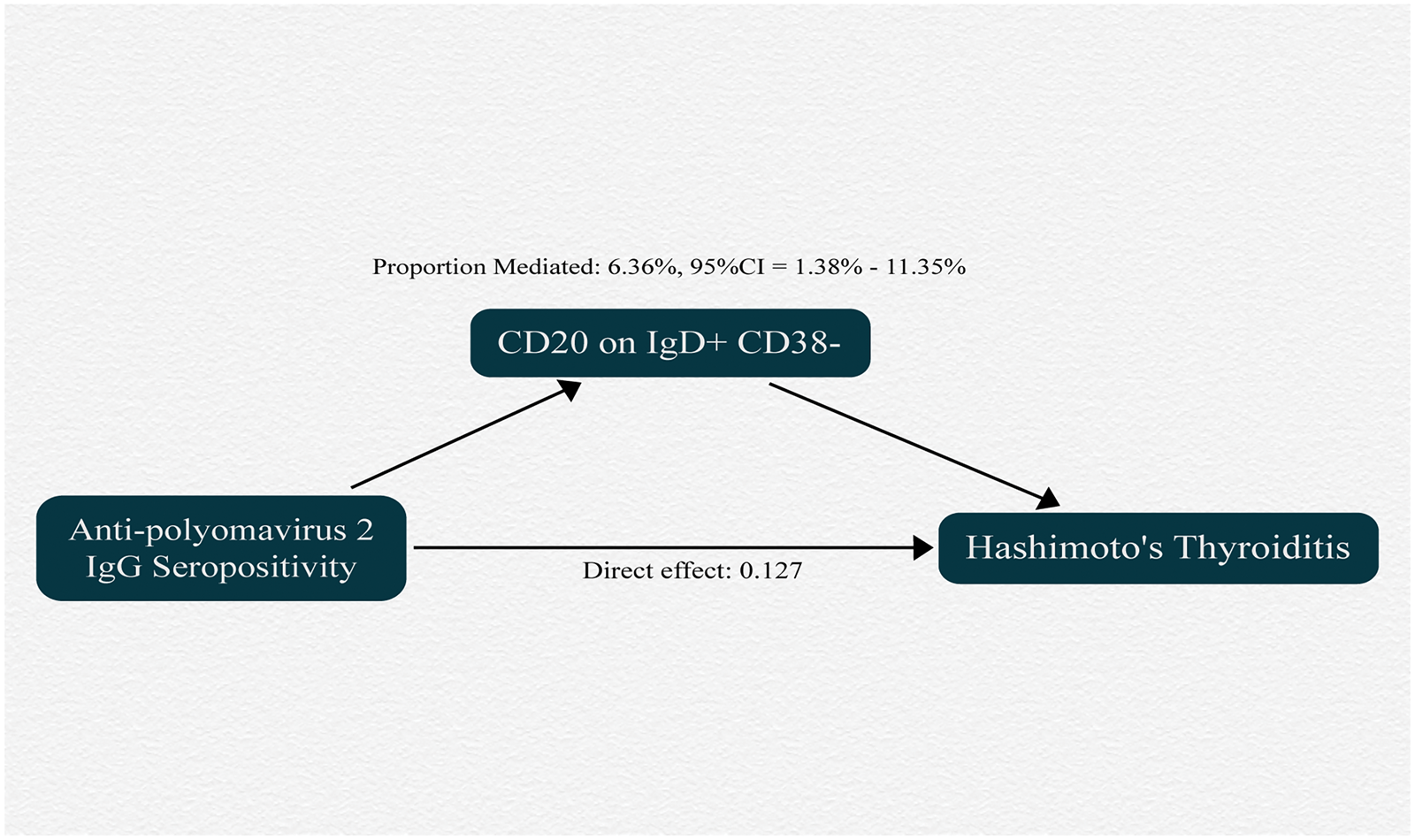
Schematic diagram showing the mediation effect of CD20+ IgD+ CD38− B cells. CI: confidence interval.
Sensitivity analysis
To assess the robustness of our findings, heterogeneity tests, horizontal pleiotropy analyses, outlier detection, and “leave-one-out” sensitivity analyses were performed for all results (Table 2). No significant heterogeneity or horizontal pleiotropy was observed across the MR analyses. Furthermore, the “leave-one-out” sensitivity analysis indicated that sequential exclusion of any single SNP did not significantly affect the MR outcomes (Supplementary Figure S1).
Heterogeneity and pleiotropy analyses of all MR studies.

SNPs: single nucleotide polymorphisms; MR: Mendelian randomization; HT: Hashimoto’s thyroiditis, IVW: inverse variance weighted; SE: standard error.
Discussion
The relationship between pathogen infections and autoimmune diseases is increasingly recognized.5,6 Current hypotheses include the following. 1. Infections stimulate the innate immune system to release cytokines and induce the expression of costimulatory factors on antigen-presenting cells, a phenomenon known as “bystander activation.” 2. Microbial peptides may share structural homology with self-antigens, supporting the widely accepted molecular mimicry theory. 3. Infection-induced local tissue damage may expose previously isolated proteins, releasing new autoantigens in a process known as “epitope spreading.” 20 Although substantial evidence suggests that pathogen infections may act as a primary trigger for thyroid dysfunction, existing findings remain fragmented and insufficient to establish a complete causal chain. 21
GWAS data from various pathogen infections were analyzed in this study and, using MR analysis, a significant causal relationship between polyomavirus 2 infection and HT was identified, with a relatively limited mediation by immune cells. In contrast, associations between HT and infections, such as the Epstein–Barr virus, hepatitis C virus, H. pylori, and HHV-6, suggested by previously reported observational studies were not supported by the present findings. As noted in most reviews, a direct link between pathogen infection and the onset of HT has not been established, and efforts to detect viruses within the thyroid tissue of patients with HT have yielded unreliable results.3,19–21 To the best of our knowledge, this study is the first to analyze the causal relationship between pathogen infection and HT. Given that such infections are generally preventable, elucidating this causal relationship can substantially improve the clinical management and treatment of patients with HT.
Polyomavirus 2, also known as BK virus (BKV), is a nonenveloped, circular double-stranded DNA virus comprising approximately 5300 bp. 48 It can be transmitted via respiratory and fecal–oral routes and is commonly found in the general population. 49 The viral capsid, with a diameter of 40–45 nm, consists of pentamers of the VP1 structural protein, forming an icosahedral structure. 50 VP1 is the only capsid protein exposed on the outer surface of the viral shell and can bind to host cell receptors, facilitating viral entry into cells. 51 Our study identified a positive causal relationship between anti-polyomavirus 2 IgG seropositivity and HT. The role of BKV in the development of urinary system–related diseases has been extensively studied.52,53 Moreover, its impact on diseases of the nervous, hematological, and respiratory systems is increasingly being explored,54,55 although the relationship between BKV and HT remains unexplored. However, existing studies have detected the VP1 gene sequence of BKV in normal and nodular thyroid tissues, 56 suggesting that the thyroid may be a potential target for BKV infection. Moreover, the presence of BKV-specific antibodies does not prevent BKV reactivation and associated pathological outcomes. 57 The viral DNA enters the cell nucleus via the nuclear pore complex, initiating replication, expression, and assembly, ultimately resulting in cellular damage, lysis, and death, along with the release of large quantities of virus and viral protein expression products. 58 This release may trigger nonspecific inflammatory responses, activating innate and adaptive humoral and cellular immunity, potentially contributing to the onset of HT. 58 Beyond HT, BKV has also been investigated in systemic lupus erythematosus (SLE) for its role in inducing autoantibodies, potentially facilitating the development of SLE.50,59
Reverse MR analysis did not reveal an inverse causal relationship between anti-polyomavirus 2 IgG seropositivity and HT, suggesting that patients with HT do not have an increased risk of BKV infection. To further investigate the role of immune cells, we conducted a mediation MR analysis across 731 immunophenotypes. Our findings suggested that BKV promotes the development of HT by modulating CD20+ IgD+ CD38− B cells; however, the indirect effect of these cells in this process was relatively weak (proportion mediated, 6.36%). CD20+ IgD+ CD38− B cells represent a specific and underexplored B-cell subset. CD38 is typically associated with cell activation and maturation, and its absence may indicate that these B cells exist in a relatively quiescent or unactivated state. CD20+ IgD+ CD38− B cells may therefore represent a more quiescent, mature subgroup of B cells that play a significant role in immune surveillance rather than immediate immune responses. This cell group may have specific functions in maintaining immune system balance and preventing autoimmune responses.
This study further contributes by examining the causal relationships between infections such as hepatitis C virus, HHV-6, H. pylori, and Epstein–Barr virus and HT. Although some fragmented evidence suggests that these pathogens may contribute to the development of HT, a complete causal chain has not been established. 21 Our findings indicate that there is no causal relationship between these pathogen infections and HT. Notably, GWAS data from two East Asian populations were used to investigate the relationship between hepatitis C virus infection and HT, and the negative results challenge previous findings.
This study has notable strengths. First, to the best of our knowledge, this is the first study to use two-sample MR analysis to comprehensively investigate the potential causal relationships between various pathogenic infections and HT and to examine the mediating role of immune cells. This approach enhances our understanding of the role of pathogenic infections in the onset of HT, which could significantly influence clinical decision-making and public health policies. Additionally, when selecting IVs for pathogen infections, we excluded SNPs potentially associated with confounding factors, such as smoking, vitamin D deficiency, postpartum depression, and the use of calcium channel blockers and diuretics. Furthermore, all outcomes in this study were validated using heterogeneity tests, horizontal pleiotropy analyses, outlier detection, and “leave-one-out” sensitivity analyses, ensuring the robustness and reliability of our findings.
This study has some limitations. First, in addressing existing controversies, we primarily used mediation MR to explore the causal relationships among multiple pathogens, immune cells, and HT. Although this method is valuable for assessing associations between exposures and outcomes, we did not investigate the underlying complex molecular mechanisms linking these factors, nor did we conduct in vivo and in vitro experiments to validate our findings, as these analyses were beyond the scope of our primary objective. Second, our study primarily relied on data related to antibody-mediated immune responses following pathogen infections and did not include information on expression of pathogens’ mRNA and proteins within thyroid tissues. Third, the study population was predominantly European, limiting generalizability to other ethnicities or populations. Fourth, the types of pathogens included were not comprehensive. Finally, despite multiple precautions, the complete exclusion of confounding factors cannot be guaranteed.
Conclusion
This study, using mediation MR analysis, identified a significant role for BKV infection in the development of HT, with CD20+ IgD+ CD38⁻ B cells acting as mediators in the pathway linking BKV infection to HT. In contrast, no causal relationships were observed between HT and more commonly studied pathogens such as HHV-6, hepatitis C virus, Epstein–Barr virus, and H. pylori. Based on these findings, incorporating screening for BKV-specific antibodies into clinical evaluations may help healthcare professionals more effectively and manage the risk of HT onset.
Supplemental Material
sj-pdf-1-imr-10.1177_03000605251406787 - Supplemental material for Unveiling the mediating role of immune cells in the link between pathogen infections and Hashimoto’s thyroiditis
Supplemental material, sj-pdf-1-imr-10.1177_03000605251406787 for Unveiling the mediating role of immune cells in the link between pathogen infections and Hashimoto’s thyroiditis by Jie Zhou, Yixin Xu, Haitao Wang, Chao Chen and Kun Wang in Journal of International Medical Research
Supplemental Material
sj-xlsx-2-imr-10.1177_03000605251406787 - Supplemental material for Unveiling the mediating role of immune cells in the link between pathogen infections and Hashimoto’s thyroiditis
Supplemental material, sj-xlsx-2-imr-10.1177_03000605251406787 for Unveiling the mediating role of immune cells in the link between pathogen infections and Hashimoto’s thyroiditis by Jie Zhou, Yixin Xu, Haitao Wang, Chao Chen and Kun Wang in Journal of International Medical Research
Footnotes
Acknowledgments
None.
Authors’ contributions
Z.J: conceptualization, data curation, formal analysis, investigation, methodology, resources, software, validation, visualization, writing—original draft, funding acquisition, and writing—review and editing; X.Y: conceptualization, formal analysis, investigation, methodology, funding acquisition, resources, supervision, validation, visualization, writing—original draft, and writing—review and editing; W.H: methodology, resources, supervision, validation, and visualization; W.K: conceptualization, formal analysis, and methodology; C.C: formal analysis and writing—original draft.
All authors gave final approval for publication and agreed to be held accountable for the work performed therein.
Data availability statement
All data and supporting information are available within the article.
Declaration of AI use
No AI-assisted technologies were used in the preparation of this article.
Declaration of conflicting interests
The authors declare no conflict of interest financial or otherwise.
Ethics
This study was based on publicly available summary data and required no ethics approval or participant consent.
Funding
This study was funded by the Jiangsu University Medical Education Collaborative Innovation Fund Project (Grant No. JDY2023018), the Medical Research Project of the Jiangsu Health Commission (Grant No. Z2021010), and the Science and Technology Development Fund Project of Xuzhou Medical University Affiliated Hospital (Grant No. ZX202508).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.