
Abstract
Objective
This current cross-sectional study aimed to investigate the demographic characteristics and molecular epidemiology of patients with Usher syndrome in the Turkish population.
Methods
Patients who were followed up with a preliminary diagnosis of Usher syndrome from various regions of the country were included in the study. After a review of patients’ medical histories, all underwent ophthalmological and otorhinolaryngological examinations. Mutations reported in previous studies in MYO7A, PCDH15, USH1C, CDH23, and USH2A were investigated using Sanger sequencing.
Results
Fourteen (29.2%) patients had Usher syndrome type 1 and 16 (33.3%) had type 2. Eighteen (37.5%) patients could not be typed. We detected mutations in MYO7A (c.1343 + 1 G > A) in seven patients (six heterozygous and one homozygous) from seven families clinically compatible with Usher syndrome type 2 (rather than Usher syndrome type 1, as was expected based on previous studies).
Conclusions
Our preliminary findings suggest that the proportions of patients with Usher syndrome type 1 and Usher syndrome type 2 in the Turkish population were almost equal, and none of our patients was clinically compatible with Usher syndrome type 3. Although previous studies reported that mutations in MYO7A rarely caused Usher syndrome type 2, in the Turkish population, MYO7A alleles may be hypomorphic and manifest as a milder phenotype in Usher syndrome compared with other populations.
Introduction
Usher syndrome (USH) is a chronic, progressive, autosomal recessive (AR) inherited disorder with retinitis pigmentosa (RP), neurosensory hearing loss, and balance problems. The condition was first described in 1914 by Charles Usher. USH accounts for 3%–6% of patients with congenital deafness, 8%–33% of RP patients, and 50% of patients who are both blind and deaf. 1 Its global prevalence is estimated between 4 and 17 per 100,000 people worldwide. 1 USH is clinically and genetically heterogeneous. The Davenport and Omenn classification (1977), 2 which is still widely used, categorizes USH into three clinical types based on the age of RP onset, severity of hearing loss, and presence of balance problems. 3 Although this traditional classification remains in use, atypical and overlapping forms have been reported across three clinical types of USH and some major genetic causes of USH. 4
Patients with USH type 1 (USH1) are born with profound congenital sensorineural hearing loss, accompanied by vestibular dysfunction. Vision loss usually begins in the first decade of life. Patients with USH type 2 (USH2) also have congenital sensorineural hearing loss ranging from moderate to severe, but vestibular function is preserved. RP typically manifests around puberty. USH type 3 (USH3) is characterized by variable onset of hearing loss, which is progressive, and vestibular problems may occur in some patients.
Six genes have been associated with USH1: MYO7A, USH1C, CDH23, PCDH15, USH1G, and CIB2 (OMIM #276900, OMIM #605242, OMIM #605516, OMIM #601067, OMIM #607696, and OMIM #614869, respectively). Mutations in three genes are associated with USH2: USH2A, GPR98, and WHRN (OMIM #276901, OMIM #605472, and OMIM #611383, respectively). Mutations in CLRN1 (OMIM #276902) have been shown to cause USH3.
Although studies have been conducted in various countries, to the best of our knowledge, no prior study has investigated both the clinical and genetic features of patients with USH in Turkey. In this study, we aimed to describe the demographic characteristics of patients with USH from different regions of Turkey who were followed up in our clinic. In addition, mutations in MYO7A, USH1C, CDH23, PCDH15, and USH2A were investigated for the first time using Sanger sequencing to reveal the molecular epidemiology of USH in the Turkish population.
Methods
Ethical compliance
This cross-sectional study received approval from the ethics committee of Şişli Hamidiye Etfal Training and Research Hospital and was conducted in accordance with the Declaration of Helsinki (1855/09.01.2018). Written informed consent for genetic testing was obtained from patients, their parents, or legal guardians. All patient details were deidentified to ensure anonymity, and the reporting of this study conforms to the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) guidelines. 5
Participants and data collection
Patients who were followed up in our clinic with a preliminary diagnosis of USH were included in the study. After a review of patients’ medical histories, all patients received genetic counseling to identify hereditary patterns. Detailed anterior and posterior segment examinations were performed as part of the ophthalmologic examination.
Subsequently, all patients were referred to the otorhinolaryngology (ear, nose, and throat (ENT)) clinic, where detailed ENT examinations and audiometric tests were performed. Hearing loss was classified according to the American Speech−Language−Hearing Association protocol: 1. Patients with a hearing loss of 45 decibels (dB) or less were classified as mild; 2. Patients with a hearing loss of 45–70 dB were classified as moderate; and 3. Patients with a hearing loss of more than 70 dB were classified as severe. All patients underwent detailed vestibular examinations as well as magnetic resonance imaging to exclude brain and inner ear malformations. Patients without neurosensory hearing loss or those who did not consent to molecular analysis were excluded from the study. 6
Molecular analysis
Molecular analysis focused on previously reported mutations in MYO7A, PCDH15, USH1C, CDH23, and USH2A, which were investigated using Sanger sequencing (Table 1). The genomic DNA of patients was polymerase chain reaction (PCR)–amplified using the primers for exons listed in Table 1 and then sequenced. Exonuclease I (3 U) and shrimp alkaline phosphatase (0.3 U) were added to 15 μL of the PCR product, and the mixture was incubated for 1 h at 37°C. This mixture was then incubated for 15 min at 80°C. The 10-μL DNA sequencing reaction mixture contained a 3.2 pmol primer, 0.6 μL of BigDye Terminator Ready Reaction Mix (Applied Biosystems, Foster City, CA, USA), and 2 μL of 5× dilution buffer (400 mM Tris–HCl, pH 9, and 10 mM MgCl2). The cycling conditions were one cycle of 96°C for 2 min and 45 cycles of 96°C for 10 s, 55°C for 10 s, and 60°C for 4 min. The resulting products were then ethanol-precipitated, and the pellets were suspended in 10 μL of deionized formamide. Sequences were obtained using an ABI Prism 3100 (Applied Biosystems, Foster City, California, USA).
Presumably pathogenic DNA variants and scanned primers.

bp: base pair.
Near Exon 12.
Statistical analysis
Statistical analyses were performed using the Statistical Package for Social Sciences (SPSS), version 26.0 for Windows (SPSS Inc., Chicago, Illinois, USA). The mean, standard deviation, and percentage were used for descriptive statistics.
Results
Demographic features and clinical results
Fifty-eight patients with hearing loss who were followed up for RP in our clinic were evaluated within the scope of this study. Ten patients were excluded from the study: neurosensory hearing loss was not detected in the ENT examination in eight patients, and two patients did not consent to molecular analysis. Forty-eight patients were included in the study. Table S1 presents the demographic features of the patients.
Molecular analysis
A mutation in MYO7A (c.1343 + 1G>A) was detected in seven patients (five males and two females) from seven families who were clinically compatible with USH2 (rather than USH1, as was expected from previous studies). In these patients, RP without vestibular disorder was most consistent with USH2. One patient carried homozygous variants, whereas six had a single allele identified (Figure 1). No mutations were detected at the analyzed sites in the other genes. None of the investigated mutations were detected in the remaining 41 (85.6%) patients.
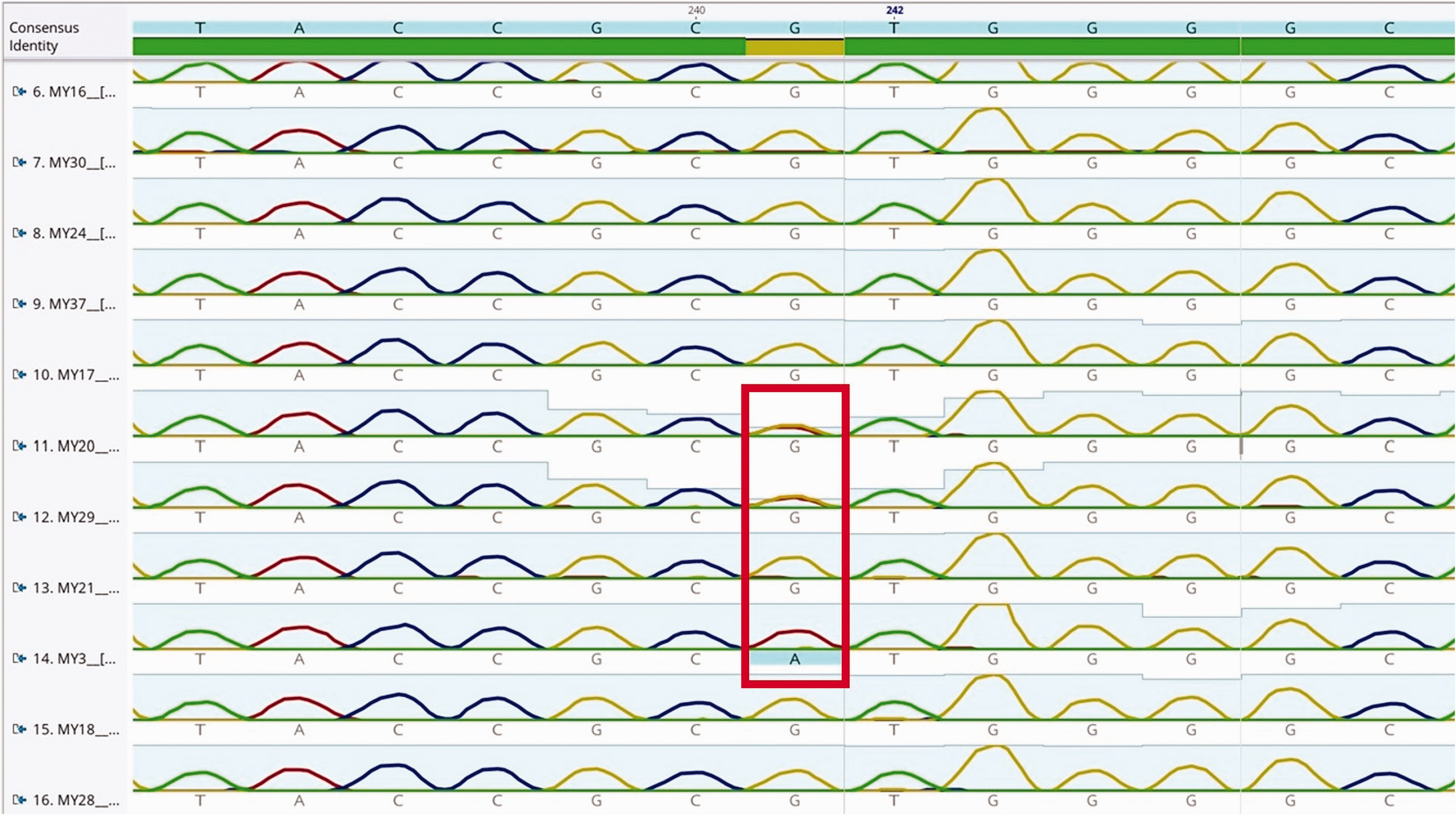
Heterozygous and homozygous mutations at the focal point of MYO7A in an image taken from Geneious Prime (Geneious version 2019.1 created by Biomatters) analysis of a group of patients.
Discussion
This study aimed to investigate the characteristics of patients with USH from various regions of Turkey who were followed up in our clinic. To the best of our knowledge, this is the first study on the demographic characteristics of patients with USH in Turkey. Fourteen patients were clinically compatible with USH1, and 16 were clinically compatible with USH2. We could not classify 18 patients. In addition, molecular analysis revealed MYO7A mutations in seven patients with USH2.
Studies from the United States and Europe have found that most patients with USH present with USH1 and USH2, while USH3 is very rare, consistent with our results.7,8 However, a study from Finland reported that the rate of occurrence of USH3 was 40%. 9 Studies from Italy, England, Germany, and China reported the prevalence of USH2 to be slightly higher than that of USH1, consistent with the findings of our study.10–13
Previous studies have demonstrated that the most common subtype of USH1 is USH1B, caused by biallelic mutations in MYO7A. Research from the United States and the United Kingdom revealed that mutations in this gene are the causative agent in approximately half of USH1 patients. Mutations in CDH23, PCDH15, USH1C, USH1G, and CIB2 also occur in patients with USH1. In 10%–35% of patients with USH1, causative mutations were identified in CDH23, and in 10% of these patients, the active mutation was detected in PCDH15. Mutations in USH2A have been identified in approximately 85% of USH2 patients.8,12,14,15
Due to budgetary limitations, we focused on MYO7A, CDH23, PCDH15, USH1C, and USH2A, which are more likely to harbor causative mutations than other genes associated with USH1 and USH2 patients. Because none of our patients were clinically classified as having USH3, mutations causing USH3 were not investigated in our study.
For mutation screening of CDH23, an 884-base sequence encoding P.R1060W aa, previously reported to harbor a mutation, was examined, but no mutation was detected.16,17 A 2016 study from England identified a mutation in PCDH15 within a 330-base sequence encoding P.R991X aa, but this mutation was not detected in our study. 18 In contrast to a 2011 French study, no mutation was found in USH1C within an 899-base sequence encoding P.R357W aa. 19 For USH2A, a 593-base sequence encoding P.G1301V aa, where a mutation was detected in a previous study in the United States, was examined, but no mutation was detected. 20
For MYO7A, a 2014 study conducted in China reported a missense mutation in c.1343 + 1G.>A. 21 Although this region is an intron, previous work has shown that it is one of the division regions of the gene, and mutations in this region can cause defects in amino acid synthesis.22,23 In our study, we analyzed a 354-base sequence covering the same region. Heterozygous mutations were detected in six patients, with a homozygous mutation in one patient. Rong et al. 21 reported biallelic heterozygous mutations, MYO7A c.(1343 + 1G>A);(2837T>G), in two patients from the same family. In our study, the c.2837T>G mutation could not be evaluated; thus, we could not determine whether the mutations we detected were biallelic. However, Rong et al. 21 found that the clinical presentation of patients with this mutation was consistent with USH2, similar to our results.
Previous studies have demonstrated that besides causing USH1, mutations in MYO7A may cause an atypical form of USH3, nonsyndromic deafness, or Leber congenital amaurosis.21,24 In addition to these known phenotypic variations, an MYO7A mutation, previously discerned in only two patients with USH2, was detected in seven patients in our study.
In Turkey, the rate of consanguineous marriage is 17% in large cities and 19% in smaller cities. In rural areas, the rate increases to 36%. 25 It has also been proven that consanguineous marriage, occurring at a global rate of 1/9, increases the incidence of hereditary diseases with AR patterns, such as USH. 26 Thus, our findings provide further evidence demonstrating the role of consanguineous marriage and highlight the importance of genetic counseling in families at risk of genetic diseases, such as USH.
This study has several limitations. Electroretinography could not be performed within the scope of our study, and visual field tests could not be conducted owing to poor patient cooperation. Although patients with USH from many regions of Turkey are followed in our clinic, a multicenter study is needed to reveal the epidemiology of the syndrome more clearly. Next-generation sequencing (NGS) is currently used for mutation screening. This technique is preferred because it yields faster results than Sanger sequencing and allows whole-genome analysis. However, Sanger sequencing is still used to confirm the findings after initial screening. Sanger sequencing remains a reliable method in terms of the accuracy of the mutations detected. 27 Moreover, we could only screen certain mutations with Sanger sequencing instead of NGS due to financial problems. We could screen only for known variants in the selected genes using Sanger sequencing due to financial limitations, indicating that novel variants, which may be unique to people of Turkish origin, were missed. Further studies are needed to clarify the molecular epidemiology of USH in Turkey.
In conclusion, the proportions of USH1 and USH2 patients in the Turkish population were similar, and none of our patients were clinically compatible with USH3. Although previous studies reported that MYO7A mutations rarely cause USH2, the results of our preliminary study suggest that patients with USH in the Turkish population may have a different mutation spectrum than that observed in other populations. To the best of our knowledge, this is the first study reporting both the epidemiological and genetic characteristics of USH in Turkey. We believe that our study yields clinically important results that may serve as the basis for further studies in this field.
Supplemental Material
sj-pdf-1-imr-10.1177_03000605251381475 - Supplemental material for Epidemiological and genetic insights of Usher syndrome in Turkish population: A cross-sectional preliminary study from University of Health Sciences, Turkey
Supplemental material, sj-pdf-1-imr-10.1177_03000605251381475 for Epidemiological and genetic insights of Usher syndrome in Turkish population: A cross-sectional preliminary study from University of Health Sciences, Turkey by Abdurrahman Alpaslan Alkan, Dilek Güven, Hakan Kaçar, Bilge Türk, Gürcan Doğukan Arslan, Gamze Karataş, Mehmet Egemen Karataş, Hani Al Saadoni, Oğuz Kaan Kutucu, Murat Karapapak, Yekta Şendül and Eyüp Düzgün in Journal of International Medical Research
Supplemental Material
sj-pdf-2-imr-10.1177_03000605251381475 - Supplemental material for Epidemiological and genetic insights of Usher syndrome in Turkish population: A cross-sectional preliminary study from University of Health Sciences, Turkey
Supplemental material, sj-pdf-2-imr-10.1177_03000605251381475 for Epidemiological and genetic insights of Usher syndrome in Turkish population: A cross-sectional preliminary study from University of Health Sciences, Turkey by Abdurrahman Alpaslan Alkan, Dilek Güven, Hakan Kaçar, Bilge Türk, Gürcan Doğukan Arslan, Gamze Karataş, Mehmet Egemen Karataş, Hani Al Saadoni, Oğuz Kaan Kutucu, Murat Karapapak, Yekta Şendül and Eyüp Düzgün in Journal of International Medical Research
Footnotes
Acknowledgment
This study was presented at the 53rd National Congress of Turkish Ophthalmological Association in Antalya, Turkey in 2019.
Authors’ contributions
Data collection and processing:
Abdurrahman Alpaslan Alkan, Dilek Güven, Hakan Kaçar, Bilge Türk, Gürcan Doğukan Arslan, Oğuz Kaan Kutucu, Gamze Karataş, Mehmet Egemen Karataş, Hani Al Saadoni, Murat Karapapak, Yekta Şendül, Eyüp Düzgün
Examinations and data sharing:
Abdurrahman Alpaslan Alkan, Dilek Güven, Hakan Kaçar, Bilge Türk, Gürcan Doğukan Arslan, Oğuz Kaan Kutucu, Gamze Karataş, Mehmet Egemen Karataş, Hani Al Saadoni, Murat Karapapak, Yekta Şendül, Eyüp Düzgün
Analysis and interpretation:
Abdurrahman Alpaslan Alkan, Dilek Güven, Hakan Kaçar, Bilge Türk, Gürcan Doğukan Arslan, Oğuz Kaan Kutucu, Gamze Karataş, Mehmet Egemen Karataş, Hani Al Saadoni, Murat Karapapak, Yekta Şendül, Eyüp Düzgün
Writing:
Abdurrahman Alpaslan Alkan, Dilek Güven, Hakan Kaçar, Bilge Türk, Gürcan Doğukan Arslan, Oğuz Kaan Kutucu, Hani Al Saadoni, Gamze Karataş, Mehmet Egemen Karataş, Murat Karapapak, Yekta Şendül, Eyüp Düzgün
Final review and approval:
Abdurrahman Alpaslan Alkan, Dilek Güven, Hakan Kaçar, Bilge Türk, Gürcan Doğukan Arslan, Oğuz Kaan Kutucu, Gamze Karataş, Mehmet Egemen Karataş, Hani Al Saadoni, Murat Karapapak, Yekta Şendül, Eyüp Düzgün
Availability of data and material
Available upon a reasonable request.
Consent to participate
Informed consent was obtained from patients or their legal guardians.
Consent for publication
Informed consent was obtained from patients or their legal guardians.
Code availability
Not applicable.
Declaration of conflicting interests
All authors declare that they have no conflict of interest.
Ethics approval
The institutional review board approved the study protocol.
Funding
This study supported by the grant 2018/090, from University of Health Sciences, Turkey.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.