
Abstract
Herein, we report the clinical and genetic features of a patient with Usher syndrome type IB to improve our collective understanding of the disorder. The patient was a teenaged boy with congenital profound hearing loss, progressive visual loss, and vestibular hypoplasia; his parents were phenotypically normal. His pure tone audiometry hearing thresholds were 100 dB at all frequencies, and distortion product otoacoustic emission was not elicited at any frequencies in either ear. Moreover, an auditory brainstem response test at 100 dB normal hearing level revealed no relevant response waves, and a caloric test showed vestibular hypoplasia. Fundus examination revealed retinitis pigmentosa and a reduced visual field. The use of high-throughput sequencing technology to screen the patient’s family lineage for deafness-related genes revealed that the patient carried a compound heterozygous pathogenic variant of MYO7A: c.541C > T and c.6364delG. This pathogenic variant has not previously been reported. Our findings may provide a basis for genetic counseling, effective treatment, and/or gene therapy for Usher syndrome.
Introduction
The World Health Organization estimates that more than 1.5 billion people worldwide—representing 20% of the world’s population—experience hearing loss. Of these, approximately 430 million people have disabling hearing loss, and this number is expected to rise to more than 700 million by 2050. 1 Approximately 50% of congenital or childhood hearing loss is hereditary. 2 Currently, there are more than 200 known deafness genes. 3 The rate of deafness gene carriers in the Chinese population is about 6.3%, and about 1 in 20 people carry deafness genes. If both parents carry the same deafness gene, the risk of having a deaf child is greatly increased. In hereditary deafness, syndromic deafness accounts for 30% of cases and non-syndromic deafness accounts for approximately 70% of cases. 4 Non-syndromic and syndromic deafness differ in that non-syndromic deafness has deafness symptoms only, whereas syndromic deafness may be accompanied by abnormal clinical symptoms related to other organ systems. 4 Studies suggest that syndromic deafness has many different phenotypes and a relatively low incidence. Usher syndrome is the most common phenotype of syndromic deafness.
Usher syndrome is the most common cause of hereditary deafness and blindness. It is an autosomal recessive hereditary disease that is characterized mainly by varying degrees of hearing impairment, accompanied by progressive visual field defects caused by retinitis pigmentosa. Some patients also present vestibular dysfunction. Usher syndrome can be divided into types I (USH1), II (USH2), and III (USH3) according to clinical features and severity. Patients with USH1—accounting for 25% to 44% of all patients with Usher syndrome—have the most severe symptoms, with severe to profound prelingual deafness, early-onset (prepuberty) retinitis pigmentosa, and vestibular dysfunction as the main manifestations. Approximately one-quarter of children with Usher syndrome also have behavioral or psychological problems. 5
Usher syndrome is highly heterogeneous; different pathogenic gene variants lead to different subtypes of Usher syndrome. There are six clear pathogenic genes in USH1: MY07A, USH1C, CDH23, PCDH 15, SANS, and CIB2. Pathogenic variants of MYO7A are most commonly reported in USH1, contributing to 5% of all USH1-related pathogenic variants. 6 One study of MYO7A and MYO15A variants in Iranian patients reported a heterozygote missense mutation (p.V2135L [c.6403G > T]) in MYO15A. 7 Moreover, pathogenic variants of MYO7A and USH2A have been identified in a patient with Usher syndrome from an Italian family. 8
Within USH1, type IB (USH1B) is an autosomal recessive inherited disease that is characterized by deafness and blindness. It is caused by pathogenic variants in MYO7A, which encodes the heavy chain of an unconventional actin-based motor protein. The starting point of the present study was the clinical diagnosis of USH1B in a patient who had a novel heterozygous pathogenic variant in the MYO7A gene. His symptoms were characterized by severe hearing loss, vestibular dysfunction, speech disorders, and retinitis pigmentosa, starting at birth.
MYO7A is located in chromosome 11q13.5; it contains 49 exons and encodes the myosin VIIA protein, which consists of 2215 amino acids and belongs to the myosin family. 9 Myosin VIIA is a motor protein with adenosine triphosphate (ATP)ase activity that binds to actin using energy generated by the hydrolysis of ATP, and moves along the myosin filament. 10 There are three important regions contained by the protein structure of myosin VIIA: 1) the motor region at the amino terminal is the functional core functional region of myosin, which is responsible for hydrolyzing ATP, generating kinetic energy, and combining with actin; 2) the regulatory region of the neck, which combines with myosin light chain to form an isoleucine–glutamine motif; and 3) the carboxyl terminal of the tail, which mainly regulates the movement of MYO7A, including the SH3 domain, two FERM domains, and two MyTH4 domains. 11 In the ear, MYO7A is mainly expressed in hair cells in the utricle and semicircular canals, and is involved in the transmembrane transport of proteins and the maintenance of hair cell function in the inner ear. In the eye, MYO7A is mainly expressed in photoreceptors and pigmented epithelial cells; its main function is the transport of visual proteins together with connecting cilia. In most cases, pathogenic variants in MYO7A result in dysfunction of the encoded protein, leading to sensorineural hearing loss and visual impairment. In the eye, the dysfunctional protein can be compensated for by other proteins in the retina. By contrast, its function in the inner ear is unique; patients with the MYO7A pathogenic variant may thus present with hearing loss, mild or no vestibular dysfunction, and the presence or absence of ocular symptoms. In the family reported in the present study, the visual, auditory, and motor symptoms were consistent with USH1B with an autosomal recessive inheritance pattern. 12
Case report
The proband was a teenaged boy with USH1B (Figure 1) from an autosomal recessive family in Jilin province, China. The proband and his family, including three individuals from two generations, were enrolled in this study: the proband’s father (I-1), his mother (I-2), and the proband himself (II-1), who had profound prelingual sensorineural hearing loss. This manuscript adheres to the applicable CARE (for CASE REports) guidelines. 13

Family pedigree of the proband (II-1) with Usher syndrome.
In the family pedigree, both parents had normal visual, auditory, and motor performance and normal visual acuity, intraocular pressure, visual field, and funduscopic examination results. Their son, the proband, had visual, auditory, and motor deficits; together, these findings are consistent with autosomal recessive inheritance characteristics. The specific manifestations of the proband were congenital profound sensorineural hearing loss and progressive vision loss. Moreover, his parents reported that the proband had delayed motor development, achieving sitting and standing abilities later than other children of the same age, and was unable to perform balance-related exercises such as running and cycling. There was no consanguinity between the proband’s parents. Furthermore, the mother was healthy during pregnancy and denied any exposure to radiation or ototoxic drugs, and the father was healthy and had no family history of related diseases.
In the proband, high-resolution images of the temporal bone under computed tomography and magnetic resonance imaging revealed no abnormalities The pure-tone hearing threshold was above 100 dB at all frequencies in both ears (Figure 2). Acoustic immittance showed a type-A curve in both ears. Distortion product otoacoustic emission showed a non-significant result; that is, the response amplitude signal-to-noise ratio was <3 dB, the detection rate was <60%, and the distortion product value was <−10 dB. The auditory brainstem also showed no significant response curve at 100 dB normal hearing level. The results of a caloric test revealed vestibular hypoplasia. Additionally, Figure 3 shows the results of a fundus examination; the patient had retinitis pigmentosa. An electroretinogram revealed severely abnormal light and dark responses in both eyes; the a- and b-wave amplitudes were significantly decreased and showed a flat pattern. Visual field examination demonstrated that the peripheral visual field was reduced and damaged in both eyes, with a tubular visual field noted. Because the proband’s symptoms and results indicated a diagnosis of Usher syndrome, the clinic recommended that he and his parents undergo genetic testing.
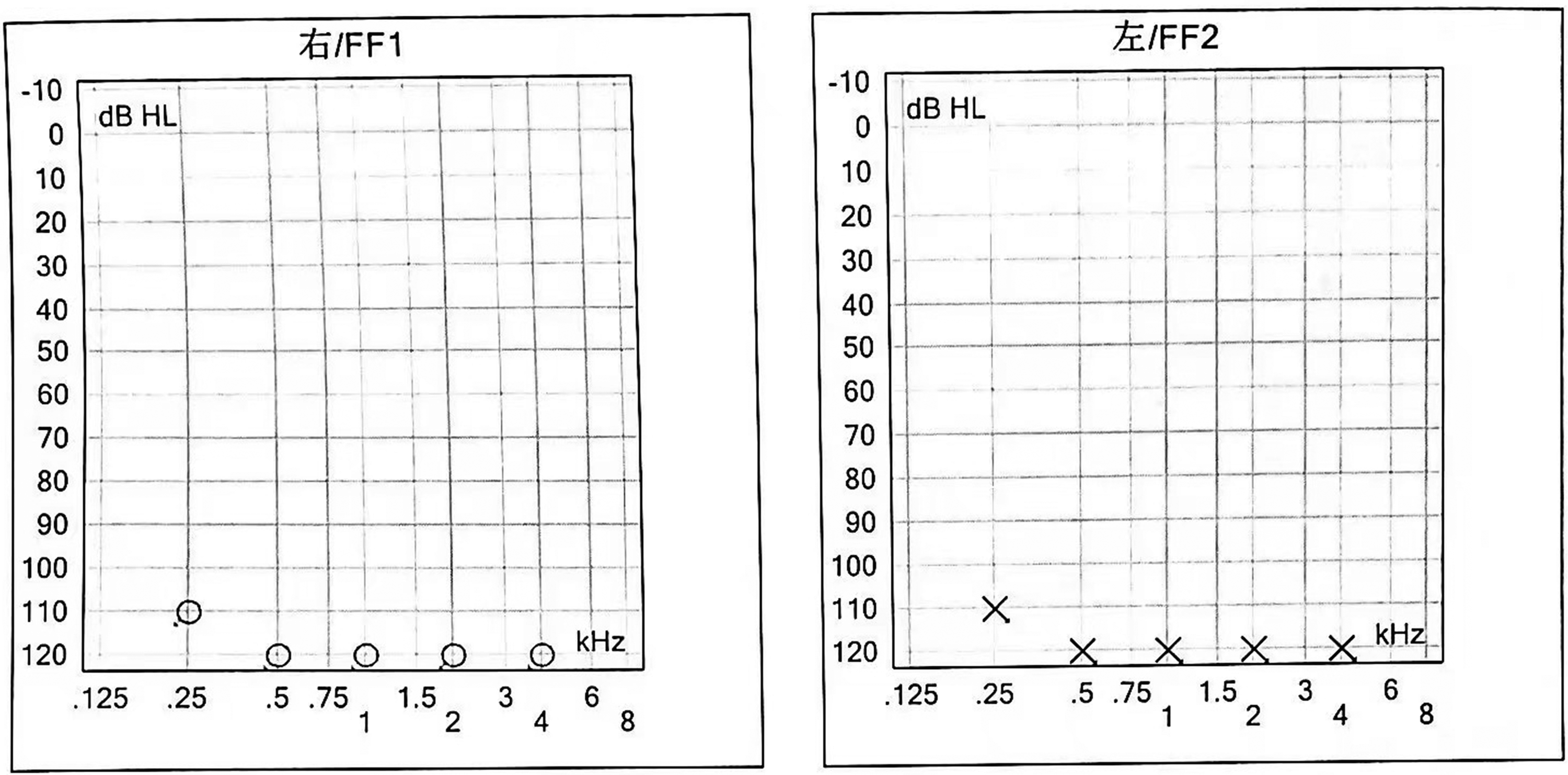
Pure-tone audiometry results of the proband. dB HL, decibels normal hearing level.

Fundal examination results of the proband. FF, Free Field (default settings).
Genetic analysis methods
After obtaining consent from the proband’s parents, the proband and his parents underwent whole-exome sequencing; Figure 4 shows the data interpretation pipeline.
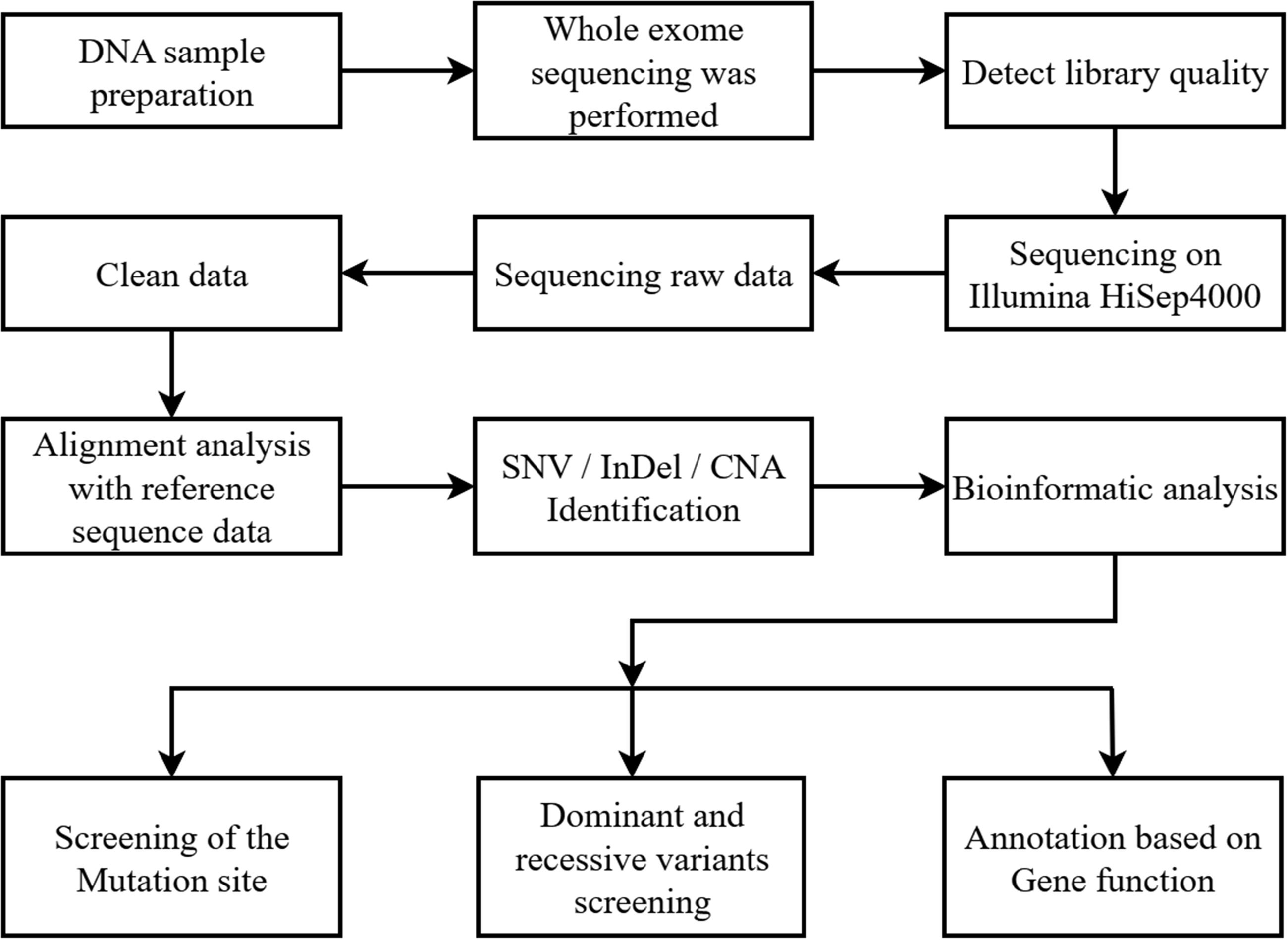
Data interpretation pipeline for whole-exome sequencing. CNA, copy number alteration; InDel, short insertions or deletions; SNV, single nucleotide variant.
Whole-exome analysis and sequencing
First, 5 mL of peripheral venous blood was collected from the patient and his family for DNA extraction and deafness gene analysis. Next, genomic DNA was extracted a using QIAamp DNA Blood Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions, and DNA concentrations were quantified (150 ng/mL).
DNA libraries were captured using the xGen Exome Research Panel v1.0 (Integrated DNA Technologies, Inc., Coralville, IA, USA). Final libraries were quantified and sequenced using a HiSeq sequencer (Illumina, Inc., San Diego, CA, USA), of 150 bp with paired ends. Single nucleotide variants and short insertions or deletions were called using the Genome Analysis Toolkit (v3.3). The variants were then merged into a Variant Call Format file with multiple samples. The single nucleotide variants and short insertions or deletions were annotated with their predicted impact against Ensembl 75, presence in the Human Gene Mutation Database (HGMD; version 2017.2) and their minor allele frequency in the Genome Aggregation Database (release r.2.0.1) using SnpEff8 4.0.
Potential pathogenic variant sites were searched in the relevant databases (HGMD, PubMed, Exome Sequencing Project, ClinVar, and 1000 Genome Project) according to the clinical phenotype of each patient. American College of Medical Genetics (ACMG) genetic variation classification criteria were used to assess variant pathogenicity.14–18 Quality control data for the whole-exome sequencing are illustrated in Table 1.
Quality control data of whole-exome sequencing

Sanger sequencing
The University of California Santa Cruz online database (https://genome.ucsc.edu/) was used to query the reference sequence near the predicted pathogenic variant site; this was used as the DNA template. The polymerase chain reaction (PCR) primers for the predicted pathogenic variant site were designed using Primer3 online primer design software (https://bioinfo.ut.ee/primer3-0.4.0/primer3/), as follows: MYO7A c.541C > T forward: 5′-GGAGGGTCCGTATTGTCAGC-3′, reverse: 5′-CCAAATGCTGCGGAGTGAGT-3′; MYO7A c. 6364delG forward: 5′-CCGTCAGTACCACATAGGCA-3′, reverse: 5′-CTGCTCACCTTCGTTTGG-3′. DNA fragments of the pathogenic variant site were amplified from the proband and his parents using quantitative PCR. Final products that met the sequencing standards were analyzed for the pathogenic variants by Sanger sequencing, and the genotype and phenotype were then analyzed in the pedigree. The Sanger sequencing primers were as follows: chr11:76867776 forward: 5′-GAAGGAGAGTGCGGTGGAG-3′, reverse: 5′-ATTGCGGATGGTCTTGGCA-3′; chr11:76924004 forward: 5′-CTTGGGCTGAGAGGGAGGA-3′, reverse: 5′-GTGGAGCAAGCGTTGGGTA-3′.
Pathogenic variant site conservation analysis
The online software T-Coffee (https://www.ebi.ac.uk/Tools/msa/tcoffee/) was used to conduct the eight-species target amino acid sequence conservation analysis. The amino acid sequence of each species was obtained from the National Center for Biotechnology Information Gene database.
Genetic analysis results
Next-generation sequencing results of the proband and parents
As shown in Figure 5 and Table 2 (referring to the GRCh38 variant of MYO7A), the next-generation sequencing analysis of the proband revealed a compound heterozygous pathogenic variant of MYO7A: c.541C > T (exon 6) and c.6364delG (exon 47). Sanger sequencing revealed that the c.6364delG pathogenic variant was inherited from the father and the c.541C > T pathogenic variant was inherited from the mother. The corresponding compound heterozygous pathogenic variant was detected in the serum of the proband.

Compound heterozygous pathogenic variant of MYO7A at locus 1/locus 2 in the proband (II-1), with co-segregation of genotype and phenotype.
Genotypes of the family line.

I-1, the proband’s father; I-2, the proband’s mother; II-1, the proband.
Prediction and conservation analysis of variant pathogenicity
The compound heterozygous pathogenetic variant c.541C > T and c.6364 delG in MYO7 A was not included in the HGMD, Exome Sequencing Project, 1000 Genome Project, ClinVar, or PubMed databases. It belongs to a recessive inherited disease (ACMG classification: PM2) that results in the following amino acid changes: p. Glu2122 fs (frameshift mutation) and p. Gln181* (termination mutation). This mutation is expected to cause the encoded protein to be truncated and lose its normal function (ACMG classification: PVS1). The mutation was therefore suspected to be pathogenic (ACMG classification: PVS1 + PM2).
Figure 6 shows the amino acid conservation analysis with T-Coffee, which indicated that c.541C > T and c.6364delG were highly homologous and highly conserved sequences in MYO7A in the human, chimpanzee, rhesus monkey, dog, bovine, rat, chicken, zebrafish and clawed frog. It therefore plays an important role in maintaining the structure and function of the protein, and the pathogenic variant may lead to structural changes and functional defects.

Conservativeness analysis of mutant site.
Figure 7 shows the two pathogenic variants of MYO7A from the present study: c.541C > T and c.6364delG. The c.541C > T variant results in a frameshift pathogenic variant of glutamic acid at position 2122, whereas c.6364delG results in termination at the glutamine at position 181. We used AlphaFold2 v2.2.0 to predict the structures of wild-type MYO7A and the two truncated mutant proteins (Figure 8), and selected the most complete BFD database in the sequence search in AlphaFold. All predictions were calculated using an Nvidia A100 graphics card, and five structures were predicted in parallel. Finally, the structures with the highest pLDDTs scores were selected for subsequent analysis in PyMOL.

MYO7A gene structure and a schematic showing the positions of its two pathogenic variants in myosin VIIA protein.

Close-up comparison of the wild-type and pathogenic variants of p.Glu2122fs and p.Gln181Ter residues at the pathogenic variant site. WT, wild type.
Diagnosis and treatment recommendations
With the results of the genetic testing, the proband was diagnosed with USH1B. Cochlear implantation was recommended to improve his hearing, accompanied by speech rehabilitation training. It was also suggested that the proband wear ultraviolet glasses to protect his eyes, actively manage his complications, take vitamin A supplements to slow the progression of retinitis pigmentosa, receive visual rehabilitation training, undergo mental and psychological treatments, receive regular visual acuity testing, and use visual aids to supplement his residual vision. Moreover, it was recommended that other members of the proband’s family undergo genetic testing before the births of future children, as well as testing to detect risk factors at an early age, so that genetic blockade can be implemented (if available).
Discussion
Hearing loss is the most common sensory deficit. It usually occurs as the result of a single gene defect, either autosomal recessive, X-linked, or mitochondrial. Usher syndrome is characterized by syndromic hearing loss. 19 The prevalence of Usher disease is 3 to 6 per 100,000 people;20,21 it accounts for 18% of all retinitis pigmentosa cases. 22
In all types of Usher syndrome, hearing loss precedes visual impairment. Balance deficits in USH1 and USH3 may be further exacerbated by vision loss as a result of retinitis pigmentosa. 23 Within each type of Usher syndrome, there are many subtypes, all of which are associated with different genes and pathogenic variants. Currently, 11 pathogenic or suspected pathogenic genes have been identified: USH1 is subdivided into USH1B, USH1C, USH1D, USH1F, USH1G, and USH1J; 24 USH2 is subdivided into USH2A, USH2C, and USH2D; 25 and USH3 is subdivided into USH3A and USH3B.
Pathogenic variants in MYO7A can cause both non-syndromic hearing loss (i.e., DFNA11 and DFNB2) and syndromic hearing loss with inner ear and retinopathy (i.e., USH1B), as well as familial Ménière’s disease. The MYO7A gene is conserved in chimpanzees, rhesus monkeys, dogs, cattle, mice, rats, chickens, zebrafish, fruit flies, and frogs. 26 In the present study, next-generation sequencing was performed on the proband and his parents. By targeting all exons and splicing regions of 531 deafness-related genes, two pathogenic variants, NM_000260: c.541C > T (p.Gln181*) and c.6364delG (p.Glu2122fs), were identified in MYO7A in the proband. A genotype analysis of the family by Sanger sequencing revealed that the pathogenic variants co-segregated with phenotype in the family. The c.541C > T (p.Gln181*) variant was located in the myosin motor domain of the myosin VIIA protein structure, whereas c.6364delG (p.Glu2122fs) was located in the FERM2 domain (Figure 7). Neither variant has been previously reported. Moreover, the variants were located in the splicing region of mRNA, and the sequence is highly conserved.
Alterations of the myosin motor domain affect the stability of information transmission and material exchange in hair cells; this affects cellular function and leads to hearing loss. The MyTH4–FERM structural domain mediates interactions between MYO7A and the Sans and Harmonin proteins. When a point pathogenic variant occurs that affects this structural domain, the translated protein loses its ability to interact with both proteins, which in turn affects its normal function. Both of the identified pathogenic variants lead to the premature termination of translation, resulting in truncated proteins with the subsequent deletion of more than 2000 amino acid residues. This large number of deletions likely has a major impact on the important function of myosin 7A proteins in connecting the cytosolic and cytoskeletal proteins. These altered connections between proteins may lead to the misfolding of amino acids, thus resulting in a loss of the entire protein function. 27
The findings from the present study expand the pathogenic variant spectrum of MY07A transcripts in Chinese patients with Usher syndrome. They are of great importance for prenatal genetic diagnosis and prevention, genetic counseling, and the future gene therapy of Usher syndrome. We also hope that the identified novel pathogenic variant site may help more clinicians to better understand Usher syndrome, thus leading to the earlier and more accurate diagnosis and treatment of patients. Given that vision gradually decreases in patients with Usher syndrome, early diagnosis can provide patients with enough time to adapt to these changes.
Supplemental Material
sj-pdf-1-imr-10.1177_03000605231218924 - Supplemental material for Identification of a novel compound heterozygous pathogenic variant in MYO7A causing Usher syndrome type IB in a Chinese patient: a case report
Supplemental material, sj-pdf-1-imr-10.1177_03000605231218924 for Identification of a novel compound heterozygous pathogenic variant in MYO7A causing Usher syndrome type IB in a Chinese patient: a case report by Ya’nan Zhang, Xinyi Guo, Ling Hao, Meihui Tian, Yuan Ma and Yong Tang in Journal of International Medical Research
Footnotes
Acknowledgements
We are grateful to the family for their willing participation and cooperation in this report. We also thank the doctors, nurses, and researchers who participated in this study.
Author contributions
Data analysis: YZ, LH. Project administration: YZ, MT. Software: YM, XG. Writing – original draft: YZ, XG. Supervision: YT.
Declaration of conflicting interests
The authors declare that there is no conflict of interest.
Ethics statement
Written informed consent was obtained from the legally authorized representative of the patient for his anonymized information to be published in this article. This case report contains no identifiable information about the patient. Ethics committee approval is not applicable because case reports do not need to be approved by a review board.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.