
Abstract
Background
Epigenetics, encompassing DNA and RNA modifications, has emerged as a prominent area of research in the post-genomic era. Numerous studies have elucidated the impact of epigenetics on tumor regulation. However, the methylation patterns of 5-methylcytosine in cervical cancer as well as its role in the tumor microenvironment and immunotherapy remain poorly understood.
Methods
Utilizing a comprehensive dataset encompassing samples from 306 patients with cervical cancer from The Cancer Genome Atlas and Gene Expression Omnibus repositories, we conducted an in-depth analysis to evaluate the potential association between the modification patterns of 5-methylcytosine and the infiltration of cells within the tumor microenvironment, taking into account 11 regulators of 5-methylcytosine modification. Subsequently, we employed stepwise regression and Least Absolute Shrinkage and Selection Operator Cox regression to quantify 5-methylcytosine modification patterns in patients with cervical squamous cell carcinoma and endocervical adenocarcinoma, yielding the 5-methylcytosine score. Our study explored the link between the 5-methylcytosine score and clinical characteristics as well as prognostic outcomes in patients with cervical squamous cell carcinoma and endocervical adenocarcinoma.
Results
A comprehensive analysis of 306 patients with cervical cancer revealed two distinct 5-methylcytosine modification patterns, henceforth labeled as 5-methylcytosine clusters A and B. These clusters exhibited distinct immunological profiles and biological attributes, with 5-methylcytosine cluster A exhibiting a higher degree of immune cell infiltration. Utilizing univariate Cox regression analysis, we identified 367 genes regulated by 5-methylcytosine that were significantly correlated with patient prognosis. This analysis further stratified the samples into three distinct genomic subtypes. Survival analyses indicated that patients belonging to gene cluster C exhibited more favorable survival outcomes than those belonging to gene clusters A and B. Intriguingly, most 5-methylcytosine regulatory factors had higher expression levels in gene cluster B than in gene cluster A. Gene set enrichment analysis of a single sample revealed elevated immune cell infiltration within gene cluster B, indicating a stronger immune response in this cluster. The 5-methylcytosine score feature was utilized to determine the 5-methylcytosine modification pattern in cervical cancer, revealing that patients with low 5-methylcytosine scores exhibited better survival rates, whereas those with high scores had increased mutation frequencies and better treatment responses.
Conclusions
This research underscores the key role of 5-methylcytosine modification patterns in cervical cancer. Analysis of these patterns will deepen our understanding of the cervical cancer tumor microenvironment, paving the way for the development of more refined and effective immunotherapy strategies.
Introduction
Globally, cervical cancer ranks as the 14th most common cancer and the fourth most prevalent malignancy among women. In 2020, the World Health Organization reported the diagnosis of approximately 604,000 new cases of cervical cancer, leading to over 342,000 fatalities worldwide. 1 Currently, the primary treatment for cervical cancer is surgery combined with chemoradiotherapy; however, the recurrence rate in advanced-stage patients can reach up to 20%.2–5 Researchers have proposed immune checkpoint targeted therapy as a new approach in treating cervical cancer, and identifying potential targets and foundations for immunocombined therapy is considered a top priority.
Ample evidence underscores the pivotal significance of epigenetics in cervical cancer progression. Among the crucial processes in epigenetics, RNA modification plays a fundamental role in regulating post-transcriptional gene expression.6–8 Specifically, 5-methylcytosine (m5C) modification, which involves the enzymatic addition of an active methyl group to the fifth carbon of cytosine in RNA, is facilitated by a donor molecule. This modification is widely observed in both messenger RNA (mRNA) and noncoding RNAs, including transfer RNA (tRNA), ribosomal RNA (rRNA), small nuclear RNA (snRNA), and enhancer RNA. The m5C modification of mRNA has been linked to various biological processes, including mRNA stability, splicing, nuclear export, DNA damage repair, and proliferation and migration of cells as well as the development, differentiation, and reprogramming of stem cells. Abnormal m5C modification of mRNA has been associated with the pathogenesis of various diseases, including atherosclerosis, autoimmune diseases, and cancer.9–12 Notably, DNA methylation, particularly at the fifth carbon of cytosine (m5C), is an epigenetic modification that plays a crucial role in regulating gene expression and cellular processes. However, although both RNA and DNA methylation involve the addition of a methyl group to cytosine, they are mediated by different sets of enzymes and contribute to distinct biological functions. The primary orchestrators of m5C modification in RNA are enzymes belonging to three distinct protein categories: (a) methyltransferases, which function as “writers”; (b) demethylases, which act as “erasers”; and (c) reader proteins, which serve as “readers” in this epigenetic process. These processes specifically regulate RNA function, whereas DNA methylation primarily influences gene expression, chromatin structure, and cellular differentiation.
Many proteins have been identified as key players in the m5C methylation process. m5C writers include the NSUN family, DNMT2, and members of the TRDMT family, which are responsible for adding the methyl group to RNA. m5C erasers, primarily the TET family of enzymes, remove the methyl group from RNA. Finally, m5C readers, such as ALYREF and YBX1, recognize the m5C modification and mediate its downstream effects, such as RNA nuclear export and stability. To uncover the intricacies of the mechanisms governing m5C RNA modification in post-transcriptional regulation, a thorough comprehension of its regulatory factors is paramount. Multiple lines of evidence have indicated a connection between aberrations in critical biological functions, including cell growth and apoptosis, embryonic stem cell renewal, cellular responses to cytotoxic stress, cancer advancement, and dysregulated immune function, and the perturbation of m5C regulatory factors.
For instance, it has been demonstrated that the m5C reader FMRP coordinates with the m5C writer TRDMT1 and the eraser TET1 to promote homologous recombination in transcriptional coupling. This coordination facilitates mRNA-dependent repair and survival in cancer cells. 13 Furthermore, NSUN2 enhances the m5C modification of GRB2, stabilizing its transcript through a GRB2-dependent mechanism. The advancement and growth of esophageal squamous cell carcinoma (ESCC) is substantially fueled by this enhancement, highlighting its crucial role in disease progression. Analysis of ESCC transcriptomes revealed an increase in m5C methylation due to the overexpression of the m5C methyltransferase NSUN2 within ESCC tumors. 14
The tumor microenvironment (TME) encompasses a diverse array of components beyond neoplastic cells alone, including stromal cells such as fibroblasts as well as immune and inflammatory cells, astrocytes, and other cellular elements that surround and interact with the tumor. Additionally, it includes the extracellular matrix, microvasculature, and infiltrating biochemical mediators. The TME plays a pivotal role in various stages of tumorigenesis, including local resistance mechanisms, immune escape strategies, and distant metastasis. Furthermore, metastasis depends on complex interactions among diverse cell types and molecular factors within the TME.
A thorough understanding of the interplay between the TME and m5C regulators may elucidate the mechanisms underlying TME immune regulation. In this study, we integrated genomic datasets from cervical squamous cell carcinoma and endocervical adenocarcinoma (CESE) samples retrieved from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) to assess the patterns of m5C modification and their correlation with the TME. This study aimed to deepen our understanding of how m5C modification contributes to shaping the TME in CESE.
Methods
Data source and preprocessing
In our study of cervical cancer, we acquired the TCGA dataset focused on CESE. This comprehensive dataset encompasses RNA sequencing, genome mutation, and clinical information, spanning three normal and 306 tumor samples. The dataset was retrieved from TCGA (http://cancergenome.nih.gov/) on 19 May 2024. Additionally, we downloaded GSE30759, comprising 63 CESE samples, from GEO (http://www.ncbi.nlm.nih.gov/geo/). We also procured somatic mutation data from TCGA, excluding samples lacking survival information. To mitigate potential batch effects, we utilized the ComBat algorithm from the sva package.
For the RNA sequencing data of TCGA-CESE, we converted the Fragments Per Kilobase Million values to Transcripts Per Kilobase Million values. Both somatic mutation and copy number variation (CNV) data were obtained from TCGA, with gene values annotated based on hg19. To visualize the CNV patterns of 24 m5C regulators across 23 chromosomes, we employed the ‘RCircos’ R package. The entire data analysis was performed using R software (version 4.3.1) and Bioconductor packages, ensuring the rigor and reproducibility of our findings.
Unsupervised clustering of m5C modifications
To delineate diverse m5C modification patterns, a comprehensive collection of 24 m5C regulators was assembled from prior research. This ensemble comprised 18 methyltransferases, also known as “writers” (NSUN1, NSUN2, NSUN3, NSUN4, NSUN5, NSUN5a, NSUN5b, NSUN5c, NSUN6, NSUN7, DNMT1, DNMT2, DNMT3A, DNMT3B, DOP2, TRDMT1, TRM4A, and TRM4B), four demethylases or “erasers” (TET1, TET2, TET3, and ALKBH1), and two “readers” (ALYREF and YBX1).
Leveraging gene set variation (GSVA) for functional annotation purposes
Utilizing a nonparametric unsupervised approach, GSVA assesses variations in pathways and alterations in the activities of biological processes within expression datasets, as reported by Hänzelmann et al. 15 . We analyzed gene enrichment and biological process enrichment between m5C modification modes using the GSVA R package. Corrected gene sets from MSigDB were downloaded for analysis. Significant enrichments were identified with a corrected p-value of <0.05. To annotate the functional roles of m5C-associated genes, we utilized the clusterProfiler R package, applying a false discovery rate threshold of <0.05 to ensure statistical significance.
Differentially expressed genes in m5C patterns
To identify genes that correlate with distinct m5C modification patterns, the patients were classified into several m5C clusters according to the expression levels of 24 m5C regulators. Subsequently, the empirical Bayesian approach implemented in the ‘limma’ R package was applied to detect differentially expressed genes (DEGs) across three distinct m5C modification profiles. 16 The threshold for statistical significance of DEGs was set at an absolute log fold change (|logFC|) of >1 and a p-value of <0.05.
Cellular infiltration of the TME
To quantify the relative abundance of immune cell infiltration within the CESE TME, we employed single-sample GSEA. As previously described by Charoentong et al., 17 we utilized gene sets specific to various immune cell types. The resulting enrichment scores served as indicators of the relative prevalence of each immune cell subtype infiltrating each sample.
m5C signature and immunotherapy correlation
The Cancer Immune Atlas (TCIA, https://tcia.at/home) provided immunophenotypic scores (IPSs) for patients with CESE, encompassing four immunogenicity-related gene groups: effector cells, major histocompatibility complex molecules, immunosuppressive cells, and immune modulators. These IPSs, ranging from 0 to 10, were calculated using z-scores, reflecting the comprehensive expression patterns across diverse cell types. Notably, elevated IPS values signify increased immunogenicity. Additionally, a correlational analysis was performed to explore the potential association between the m5C score and the TME.
Results
Genetic variations in m5C regulators within CESE
In this study, 18 writers, 4 erasers, and 2 readers were evaluated to explore the roles of 24 m5C regulatory genes in cancer.
Initially, we investigated 24 regulators of m5C modification in cervical cancer (Figure 1(a)). In our analysis, each vertical column corresponded to an individual patient. Among the 289 samples examined, 39 displayed genetic alterations in these 24 m5C regulators, accounting for 13.49% of the total samples. Notably, TET1 and DNMT1 exhibited the most frequent mutations, at a rate of 3%, whereas TET3 and DNMT3A followed closely with a rate of 2%. In contrast, NSUN2, NSUN7, TET2, and DNMT3B displayed a more modest mutation rate of 1% (Figure 1(a)).

Genetic variation of m5C regulators in cervical cancer. (a) Mutation frequency of 24 m5C regulators in 289 patients with cervical cancer from TCGA-CESE. (b) CNV frequency of the m5C regulators analyzed. (c) CNV locations mapped on 23 chromosomes from TCGA data. (d) Significant differences in m5C regulator expression between normal and tumor samples. (*p < 0.05, **p < 0.01, ***p < 0.001; ns, not significant). TCGA: The Cancer Genome Atlas; CESE: cervical squamous cell carcinoma and endocervical adenocarcinoma; CNV: copy number variation; m5C: 5-methylcytosine.
In terms of CNVs, NSUN2, DNMT3B, ALYREF, YBX1, NSUN4, and DNMT3A showed a predisposition toward amplification, whereas NSUN3, ALKBH1, TET3, and NSUN7 displayed a higher likelihood of CNV deletions (Figure 1(b)). Figure 1(c) further illustrates the chromosomal locations where CNVs were observed in m5C regulators. Specifically, YBX1, NSUN4, DNMT3A, NSUN2, NSUN5, NSUN6, TRDMT1, TET1, ALYREF, and DNMT3B exhibited a concentrated pattern of amplification, whereas TET3, NSUN3, NSUN7, TET2, ALKBH1, and DNMT1 displayed a broader distribution of CNV deletions.
To thoroughly elucidate the complex interplay between genetic variations and the transcriptional patterns of m5C regulators, we performed an in-depth comparative analysis of the mRNA expression profiles of 24 crucial m5C regulators in both tumorous and adjacent nontumorous tissue samples. Our findings strikingly revealed a substantial upregulation in the expression of DNMT1, DNMT3B, NSUN2, NSUN5, TET3, and ALYREF within tumor tissues, whereas TRDMT1 expression alone displayed a conspicuous downregulation. This profound disparity in expression patterns suggests a pivotal regulatory role of these m5C regulators in the complex machinery of tumorigenesis. The significance of our discoveries is profound, as they provide novel perspectives on the molecular underpinnings that govern the impact of genetic variations on m5C-regulated gene expression in the cancer milieu. This knowledge potentially serves as a cornerstone for developing targeted therapeutic strategies and precision medicine approaches (Figure 1(d)).
Patterns of m5C methylation modulation by 24 key regulators
Clinical data were incorporated into the TCGA dataset to assess the prognostic potential of 32 pivotal m5C regulators in patients with cervical cancer. Our univariate Cox regression analysis revealed significant prognostic implications for these regulators, as summarized in Supplementary Table 1. The constructed m5C regulatory network underscores their prognostic importance in cervical cancer, as illustrated in Figure 2(a). Notably, the expression patterns of m5C regulators exhibited robust correlations within their respective functional classes as well as across the categories of erasers, readers, and writers. These discoveries imply that the intricate interplay among erasers, readers, writers, and m5C modification patterns holds a pivotal role in cervical cancer progression.
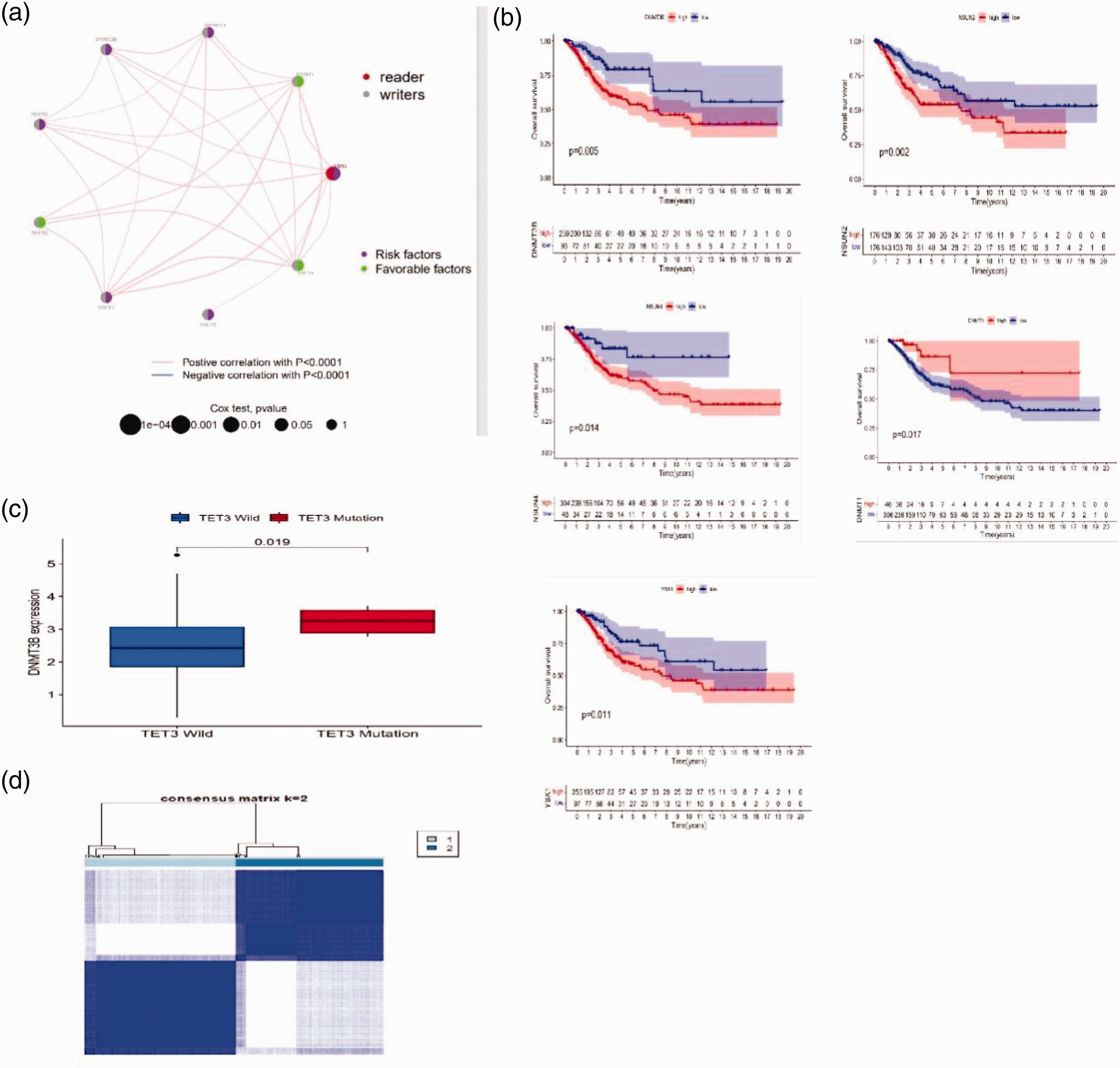
Identification of distinct m5C modification patterns via analysis of m5C regulators. (a) Identification of the network of m5C regulators in CESE. (b) Heatmap of two m5C modification patterns using 24 regulators. (c) KEGG pathway analysis. (d) Survival analysis of m5C regulators in cervical cancer using TCGA and GSE30759 datasets. (e) DNMT3B expression in TET3 wild-type and mutants. CESE: cervical squamous cell carcinoma and endocervical adenocarcinoma; TGCA: The Cancer Genome Atlas; m5C: 5-methylcytosine.
After analyzing the expression patterns of m5C regulators, we segregated the TCGA cohort into two distinct clusters: (a) m5C cluster A, encompassing 186 cases, and (b) m5C cluster B, comprising 181 cases. These clusters are depicted in Figure 2(b) and 2(c).
To delve deeper into the clinical relevance of m5C regulators in patients with CESE, we conducted an analysis of the intricate interaction network among these regulators and its influence on CESE prognosis. Interestingly, the network revealed positive correlations among all regulators, indicating a potential cooperative mechanism. Specifically, regulators including NSUN6, DNMT1, and NSUN3 were significantly associated with an unfavorable prognosis among patients with CESE. Conversely, YBX1, DNMT3B, DNMT3A, NSUN2, NSUN4, and NSUN5 displayed positive associations with favorable prognosis in CESE. These findings offer profound insights into the intricate functions of m5C regulators in CESE prognosis, thereby identifying potential therapeutic avenues for future exploration.
To assess the prognostic importance of 25 m5C regulators in CESE, we conducted a univariate Cox regression analysis, quantifying the hazard ratio (HR) associated with each regulator. Notably, YBX1 emerged as a risk factor, exhibiting a statistically significant correlation with CESE prognosis (p = 0.01011, HR = 1.861, 95% confidence interval: 1.159–2.987). To gain a deeper understanding of the prognostic value of the remaining 24 m5C regulators, we performed a Kaplan–Meier survival curve analysis.
Our analysis of data from the TCGA and GSE30759 cohorts uncovered significant associations between the expression levels of DNMT1, DNMT3B, NSUN2, NSUN4, and YBX1 and the clinical outcomes in patients with CESE (Figure 2(d)). These findings suggest that the expression patterns of these m5C regulators serve as valuable prognostic markers for CESE, offering potential insights into disease progression and therapeutic response.
In an effort to elucidate the intricate interplay between genetic mutations in m5C regulators and their expression patterns, we conducted a comprehensive analysis. Specifically, we focused on the mutation frequency of four key m5C modification factors: TET1, DNMT1, TET3, and DNMT3A. Following this, we systematically compared the expression levels of these regulators in tissues harboring wild-type genes with those harboring mutant genes. Our findings revealed significant alterations in the expression patterns of m5C regulators. Specifically, NSUN2 expression was notably upregulated in tumor tissues with NSUN7 and TET3 mutations, compared with wild-type tissues. Similarly, increased DNMT3B expression was observed in tumor tissues with NSUN7 and TET3 mutations. Conversely, DNMT3A expression was significantly downregulated in tumors harboring TET2 mutations, compared with wild-type tumors. These findings provide valuable insights into the complex regulatory network of m5C regulators and their potential role in tumorigenesis (Figure 2(e)).
Analysis of GSVA and cellular infiltration patterns in diverse m5C modification patterns
Based on the expression profiles of 24 m5C regulators, patients diagnosed with CESE were classified into two distinct clusters exhibiting varied m5C modification signatures, leveraging the ConsensusClusterPlus R package. The consensus matrix was optimized at a value of 2, exhibiting the highest efficiency and minimal crossover among the CESE samples (Figure 3(a)). Subsequently, principal component analysis was performed to delineate these two groups along principal components 1 and 2, labeled as m5C cluster A and m5C cluster B, respectively. Additionally, a heatmap was constructed to visually represent the gene expression patterns of these m5C regulators within the two clusters (Figure 3(b)). Upon analysis, we observed significantly augmented expression levels of the 24 m5C regulators in m5C cluster A, as opposed to those in m5C cluster B. However, the prognosis between patients within the m5C cluster groups exhibited no noteworthy discrepancies.

GSVA enrichment analysis and characteristics of tumor microenvironment cell infiltration in two m5C modification modes of cervical cancer. (a) Consensus clustering matrix at k = 2. (b) Principal component analysis between Cluster A and Cluster B scoregroups in cervical cancer. (c) GO enrichment of m5C DEGs. (d) KEGG pathways of m5C DEGs. GSVA: gene set variation; GO: Gene Ontology; DEGs: differentially expressed genes; KEGG: Kyoto Encyclopedia of Genes and Genomes; m5C: 5-methylcytosine.
A comprehensive analysis of GSVA was performed to gain insights into the complex biological phenotypes displayed by the two distinct clusters of m5C modification patterns, as illustrated in Figure 2(b) and 2(c). Subsequent Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis revealed considerable differences in the metabolic and signaling patterns between these two clusters. Specifically, m5C cluster A displayed a positive correlation with an array of pathways, including lysine degradation, folate-mediated one-carbon pool maintenance, ubiquitin-mediated proteolysis, RNA degradation, spliceosome function, base excision repair, homologous recombination, glycosylphosphatidylinositol-anchor biosynthesis, and selenoamino acid metabolism. These pathways primarily center around the processing, maintenance, and integrity of genetic information.
In contrast, m5C cluster B exhibited a positive correlation with a diverse array of pathways, encompassing phenylalanine metabolism, peroxisome proliferator-activated receptor signaling, steroid hormone biosynthesis, arachidonic acid metabolism, linoleic acid metabolism, neuroactive ligand–receptor interactions, calcium signaling, complement and coagulation cascades, cell adhesion molecules, hematopoietic cell lineage, and cytokine–cytokine receptor interactions. These findings suggest that m5C cluster B is enriched in pathways that are often activated in cancer, stromal cell activation, and immune responses.
In summary, this comprehensive analysis highlights the contrasting roles of the two m5C clusters, with cluster A predominantly linked to genetic information processing and cluster B associated with cancer, stromal, and immune activation pathways.
To further delineate the genetic signatures of distinct m5C clusters, we initially employed the ‘limma’ R package to identify 4753 DEGs that were pertinent to the m5C phenotypes (as depicted in Figure 3(c)). Subsequently, we conducted KEGG and Gene Ontology (GO) enrichment analyses to ascertain the biological processes encompassed by these genes. As illustrated in Figure 3(d), these genes exhibited enrichment in various immune activation-associated pathways.
With regard to GO analysis, the key biological processes were centered on histone modification, chromatin remodeling, regulation of chromosome organization, and modification of peptidyl-lysine. The analysis of cellular components revealed that the DEGs were primarily associated with SWI/SNF superfamily-type complexes, adenosine triphosphatase complexes, and transferase complexes, particularly those involved in the transfer of phosphorus-containing groups. Furthermore, molecular functional analysis highlighted the enrichment of DEGs in transcription coactivator activity, RNA polymerase II-specific DNA-binding transcription factor binding, protein serine kinase activity, and adenosine triphosphate hydrolysis activity.
In addition, KEGG enrichment analysis revealed several signaling pathways, including cell cycle, cellular senescence, animal autophagy, ubiquitin-mediated proteolysis, base excision repair mechanisms, and insulin signaling cascade.
Distinctive patterns of GSVA and TME cell infiltration in varying m5C modification modes
To delve deeper into the mechanisms underlying these disparities, we conducted unsupervised clustering analysis of the DEGs. Through univariate Cox regression analysis, we identified 367 DEGs that had a significant impact on prognosis. Based on these 367 genes, we classified patients with cancer into three distinct genomic subtypes: gene cluster A (93 cases), gene cluster B (234 cases), and gene cluster C (40 cases), as depicted in Figure 4(a). Further survival analysis revealed that gene cluster C exhibited more favorable survival outcomes than gene clusters B and A, as shown in Figure 4(b).

m5C gene patterns mediated by m5C phenotype-related DEGs. (a) Pearson correlation analysis revealed m5C regulator relationships, selecting a k = 3 clustering matrix. (b) Kaplan–Meier curves showed survival differences across m5C gene patterns. (c) Analysis of 24 m5C regulators, significance indicated by asterisks. (d) Heatmap highlights distinct m5C gene patterns. (e) Alluvial diagram showing m5C score, clusters, genes, and survival correlations and (f) m5C scores differed significantly among three gene patterns. DEGs: differentially expressed genes; m5C: 5-methylcytosine.
Additionally, the expression patterns of m5C regulators, except for NSUN5, revealed that gene cluster A exhibited elevated levels compared with gene clusters B and C, with gene cluster C displaying the lowest expression among the three (Figure 4(c)). A heatmap showed the distinct clinicopathological features of these patterns (Figure 4(d)). The expression patterns of m5C regulators was the highest in cluster A and lowest in cluster C. The 24 m5C regulators displayed significant disparities in their expression profiles across the three distinct m5C-modulated genomic subtypes (Figure 4(d)).
Identification and functional annotation of m5C gene signatures
To achieve a more nuanced prediction of m5C modification patterns at the individual patient level, we formulated a scoring system termed the m5C score. This system allows for a more precise assessment compared with group-level analysis. To visualize the characteristic alterations in patients with cervical cancer, we employed alluvial plots (Figure 4(e)). Notably, the majority of patients in m5C cluster C were from clusters B and C, whereas all patients in m5C cluster C belonged to the low-score group, with most of them showing high survival rates. Collectively, our research demonstrates the potential of m5C score as a reliable indicator for evaluating immune cell infiltration and performing prognostic evaluations, thus providing valuable insights for clinical applications.
Subsequently, we conducted a comparative analysis to examine the fluctuations in m5C scores across distinct m5C and gene clusters. Our results revealed that the m5C scores of both m5C cluster A and gene cluster A were substantially elevated compared with those of m5C cluster B and gene cluster B, respectively (Figure 4(f)). Notably, the m5C score associated with gene cluster C was significantly diminished compared with the scores observed in gene clusters A and B.
Derivation and clinical association assessment of the m5C score in cervical cancer
To further elucidate the prognostic significance of m5C score, a survival analysis of two distinct m5C score subgroups was conducted. The results revealed a significant survival advantage for patients with CESE who had low m5C scores (p < 0.001, Figure 5(a)). The mortality rate was notably higher in the low-m5C score subgroup (16%) than in the high-m5C score subgroup (Figure 5(b) and (c)). Stratified analysis further indicated that patients with low m5C scores, regardless of the Federation of Gynecology and Obstetrics G1-2 or G3-4 status, exhibited a more favorable prognosis compared with those with high m5C scores (Figure 5(d)). Additionally, we assessed the potential of m5C score as an independent prognostic biomarker for CESE.

The m5C gene signature and its role in immunotherapy. (a) Patient survival proportions vary between low- and high-m5C score subgroups. (b) Tumor m5C score was positively correlated with TMB. (c) Kaplan–Meier curves showed survival differences by m5C score. (d) Kaplan–Meier curves compared G1-2 survival with G3-4 survival. (e) The relationship between IPS and the m5C scoring system, encompassing IPS-PD1, IPS-CTLA4, and IPS-PD1/CTLA4, was examined in both low- and high-m5C score subgroups. (f) The m5C score was correlated with immune cell infiltration in the TME, indicated by blue (negative) and red (positive). TMB: tumor mutation burden; IPS: immunophenotypic scores; m5C: 5-methylcytosine.
Distinct immunotherapy profiles characterized by M5C score signatures
Approved drugs targeting PD-1 and CTLA-4 have revolutionized cancer therapy across various types. To enhance precision in immunotherapy, we delved into the correlation between m5C score signatures and IPSs in patients with cervical cancer, aiming to predict their responsiveness to immune checkpoint inhibitors. We obtained the data from TCIA and analyzed m5C modification as well as its association with immunotherapy. Our analysis revealed that patients with CESE and low m5C score who received PD-1 or CTLA-4 or combined immunotherapy had a better survival prognosis (Figure 5(e)). Furthermore, we analyzed the differential expression of immune checkpoint molecules in patients with cervical cancer stratified by high and low-m5C score groups (Figure 5(f), Supplementary Materials). This analysis revealed a strong association between m5C score and the potential response to immunotherapy.
Features of the m5c molecular subtype and tumor somatic mutations
Recent investigations have revealed a compelling correlation between genetic alterations within tumor genomes and the efficacy of immunotherapy, indicating a potential link between somatic mutations and the patient’s response to such treatment modalities. Upon analysis, we observed no substantial variance in tumor mutation burden (TMB) across various m5C score subgroups, thus indicating the lack of a direct association between m5C score and TMB (p = 0.96; Figure 6(a)). Based on TMB expression, patients with CESE were stratified into two distinct subgroups: (a) those with high TMB and (b) those with low TMB. Notably, individuals belonging to the high TMB subgroup demonstrated a more promising prognosis compared with those belonging to the low TMB subgroup. (Figure 6(b)). The integration of m5C score and TMB has resulted in refined risk stratification, revealing substantial differences in prognosis among the various m5C score subgroups (p < 0.001; Figure 6(c)). Notably, the survival analysis incorporating both TMB and m5C score highlights a significant survival benefit for patients belonging to the low-score group.

Characteristics of cervical cancer subtypes related to m5C and mutations. (a) Stratification of patients with cervical cancer by m5C score and TMB. (b) Survival analysis based on TMB. (c) Survival curve combining TMB and m5C score. (d, e) Mutation frequencies in high- and low-m5C score subgroups. TMB: tumor mutation burden; m5C: 5-methylcytosine.
Additionally, we delved into the disparity in somatic mutation distribution between the high- and low-m5C score subgroups of the TCGA-CESE dataset. As shown in Figure 6(d) and 6(e), the high-m5C score subgroup exhibited a slightly elevated somatic mutation rate compared with the low-m5C score subgroup, albeit without reaching statistical significance (84.57% vs. 84.4%). In both subgroups, TTN was identified as the most common mutation, followed by PIK3CA. These findings revealed a potential interplay between somatic mutations and individual tumor patterns of m5C methylation modification.
Discussion
Previously, m5C has been detected in the mRNA, rRNA, and tRNA of various species.18,19 As a reversible epigenetic modification,20,21 m5C modification of RNA affects the fate and function of modified RNA molecules, playing a pivotal role in a wide range of biological processes such as the regulation of RNA stability, protein–RNA interactions, and transcriptional regulation.22–24 Additionally, extensive research has highlighted the crucial role of m5C regulators in diverse biological and pathological processes, encompassing cell proliferation and differentiation, stem cell fate specification, embryonic development, tumor initiation, tumor progression, and tumor-related immune responses.18,25–27 Although research on m5C methylation patterns in cervical cancer is still in its early stages, 28 existing studies suggest that m5C modification is closely associated with the onset and progression of cervical cancer. Specifically, changes in the expression of m5C regulatory factors, such as NSUN family proteins and DNMT2, have been found to be linked to cancer metastasis, cell proliferation, and drug resistance. However, the relationship between immune infiltration in the cervical cancer TME and patterns of m5C methylation modification remains incompletely understood. The key objective of this research was to establish the m5C score, based on 24 distinct m5C methylation patterns, and assess its potential association with the immunological landscape of the TME in cervical cancer.
A contemporary scientific study has underscored the fundamental importance of m5C modification in the pathogenesis of human bladder urothelial carcinoma. Zhang et al. revealed that the enzymes NSUN2 and YBX1 are the primary mediators of methylation in the 3′-untranslated region of HDGF, thereby contributing to the progression of bladder urothelial carcinoma. Notably, elevated expression levels of NSUN2, YBX1, and HDGF have been correlated with poor survival outcomes. 29 In the context of colon adenocarcinoma, NSUN6 has been identified as a regulator that upregulates the expression of the oncogene METTL3 and catalyzes its m5C modification. Notably, the overexpression of METTL3 can significantly attenuate cell cycle arrest induced by NSUN6 deficiency, thereby inhibiting tumor development. 30 Recent investigations have underscored the importance of NSUN2 overexpression as a stand-alone predictor of the risk of hepatocellular carcinoma. This is attributed to its strong association with immune cell infiltration, malignant progression, and adverse prognosis. The suppression of NSUN2 expression results in substantial decreases in the mRNA levels of GRB2, RNF115, AATF, ADAM15, RTN3, and HDGF, thereby modulating the RAS signaling cascade, triggering cell cycle arrest, and augmenting the responsiveness of liver cancer cells to sorafenib treatment. 31 Furthermore, the expression of NSUN2 is notably upregulated in colorectal cancer, where it functions in an oncogenic manner through the NSUN2/YBX1/mC-ENO1 signaling axis. This axis exhibits a positive feedback loop and provides new insights into the therapeutic potential of NSUN2 inhibitors in combination with immunotherapy for colorectal cancer. 32 NSUN2 promotes the oncogenic activity of gastric cancer by directly interacting with SUMO-2/3, stabilizing its own structure, and mediating its nuclear translocation. 33 These previous studies investigated the role of surface RNA modifications in cancer promotion and tumor suppression.34,35
NSUN2, a key methyltransferase in the m5C modification process, plays a crucial role in adding methyl groups to RNA, thereby regulating various functions such as RNA stability and translation efficiency. A study by Wang et al. in 2022 highlighted that the upregulation of NSUN2 expression in cervical cancer is closely associated with the malignant characteristics of the disease, particularly tumor proliferation and migration. 36 The study emphasized the role of NSUN2, an m5C writer, in the mechanisms of cancer cell proliferation and metastasis, suggesting that NSUN2 not only plays a significant role in the development of cervical cancer but may also serve as a novel target for prognosis prediction and targeted therapy. Conversely, YBX1 is a reader of m5C modification, capable of accurately recognizing RNA modified with m5C and regulating the stability and translation of these RNAs, thereby affecting the growth, migration, and invasiveness of cancer cells. A study by Zheng et al. in 2023 demonstrated that circ_0002762 regulates miR-375, which further activates YBX1, leading to more pronounced malignant features in cervical cancer cells. 37 This study revealed the critical role of YBX1 in cervical cancer, demonstrating that YBX1 is not only an m5C “reader” but also promotes the malignant progression of cervical cancer by influencing cell proliferation, migration, and invasion. A study by Chen et al. in the same year further explored the role of YBX1 in cervical cancer. 38 They found that YBX1 stabilizes mRNA harboring m5C modifications, particularly LRRC8A, which supports cancer cell survival and tumorigenesis. Upregulation of LRRC8A inhibits apoptosis and promotes tumorigenesis, providing new evidence regarding the importance of YBX1 as an m5C “reader” in tumor progression. These studies highlight the distinct roles of NSUN2 and YBX1 in cervical cancer. NSUN2, an m5C writer, affects cancer cell proliferation and migration, whereas YBX1, a reader, regulates mRNA stability and function, influencing tumor progression. These findings offer valuable insights into m5C modification in cervical cancer and suggest new therapeutic strategies.
Utilizing the curated TCGA-CESE dataset, we conducted a thorough examination of CNVs within 24 regulatory coding genes that play a pivotal role in the m5C modification pathway. This comprehensive analysis aimed to further elucidate the significance of these alterations in the context of m5C modulation. Our findings indicated CNV alterations in a substantial proportion of these m5C regulators. Furthermore, through rigorous statistical analysis, we identified two distinct patterns of m5C methylation modifications, each exhibiting remarkably different immune cell infiltration characteristics within the TME. These patterns were delineated based on the expression profiles of the abovementioned 24 modulatory genes involved in m5C methylation.
Importantly, our analysis distinguished three unique genomic manifestations of the m5C gene derived from a robust collection of >4500 DEGs that exhibited significant correlations with the m5C phenotype. These findings reinforce the pivotal importance of m5C modifications in molding the TME and its intertwined immune response. Importantly, our analysis suggests that specific genes, including DNMT1, DNMT3B, NSUN2, NSUN4, and YBX1, hold significant prognostic implications for cervical cancer. This insight has the potential to inform future research on the development of targeted therapeutic strategies for this malignancy.
The intricate link between m5C modification and immune cell infiltration within the TME has been thoroughly explored. Notably, Chen et al. uncovered two distinct signatures of m5C methylation, exhibiting diverse immune cell infiltration patterns and prognostic implications, based on an array of 15 m5C regulatory factors. 39 The established m5C score further confirmed the crucial role of m5C modification in regulating lung adenocarcinoma via the TME, potentially guiding the development of more effective immunotherapy strategies. 31 Notably, most m5C RNA methylation tuning factors are differentially expressed and possess prognostic value. Moreover, there is a distinct correlation between immune infiltrating cells and the regulatory factors of m5C modification, particularly NSUN7. Xiao et al. identified SEPT3, CHI3L1, PLBD1, PHYHIPL, SAMD8, RAP1B, B3GNT5, RER1, PTPN7, SLC39A1, and MXI1 as predictive factors in gliomas and constructed prognostic signatures utilizing these genes. 40 This research further underscores the significance of m5C-related biomarkers in predicting the survival prospects of patients with glioma, enhancing our understanding of the clinical implications of these modifications.
Yu et al. discovered that TET3 is upregulated in prostate cancer, which correlates with poor prognosis. They further developed a prognostic model designated as m5C, which integrated NSUN2, TET3, and YBX1 for practical clinical use. Moreover, they succeeded in distinguishing various m5C gene clusters and immune subtypes, demonstrating notable variations in the expression patterns of regulatory genes. 41 In bladder cancer, Yan analyzed 412 patients and identified two m5C patterns associated with the TME and survival. NSUN6 inhibits M2 macrophages through m5C methylation. Our m5C score predicts better prognosis, highlighting m5C’s role in TME modulation and immunotherapy. 42 Chen identified three m5C phenotypes in 1792 patients with colorectal cancer. The m5C score predicts the immunotherapy response, negatively correlating with immune cells and PD-L1 but positively with cancer mutations. Lower m5C scores indicate better immunotherapy responses. 43 Previous studies have revealed that RNA m5C modification plays an important role in the TME and in response to immunotherapy.44,45
In our investigation, we observed a negative correlation between the m5C score and PD-L1 expression, whereas a positive association was noted between the m5C score and TMB. This underscores the pivotal role of methylation patterns in shaping the intricate immune landscape of the TME. Our results indicate that m5C modifications hold potential in influencing the therapeutic responsiveness to cancer immunotherapy in patients with CESE. Consistent with prior studies, checkpoint immunotherapy responses are not solely determined by antigen processing; they are also influenced by factors such as angiogenesis suppression, TGF-β pathway components,46–49 and epithelial-to-mesenchymal transition.48,50–55 Consequently, we propose a refined approach to modify the immune microenvironment, aiming to personalize precision immunotherapy for CESE. We hypothesized that a comprehensive assessment integrating m5C status with biomarkers such as PD-L1 expression, mutational load, and immune TME characteristics can serve as a promising predictive tool for evaluating immunotherapy efficacy.
Despite the significance of our findings, our study had certain limitations that need to be addressed. First, the use of a publicly available dataset, albeit comprehensive, lacked clinical validation. The incorporation of clinical data verification could significantly enhance the accuracy and reliability of our results. Second, further validation of the biological mechanisms underlying the role of m5C regulators in cervical cancer is imperative. Finally, owing to time constraints and budgetary limitations, we could not conduct the necessary experiments to fully validate our findings. In future studies, we plan to conduct additional experiments to validate and expand upon our current results. Although preliminary studies suggest that m5C methylation play a role in post-transcriptional regulation in cervical cancer, the current research has not yet revealed the correlation between m5C modification and different clinical stages of cervical cancer. In particular, the changes in m5C methylation levels at different stages and their impact on patient prognosis remain unclear. Additionally, the interactions between m5C methylation and other RNA modifications (such as m6A and m1A) and their combined effects in cervical cancer require further investigation.
Conclusions
Future research should focus on several aspects. First, large-scale clinical sample analysis should be conducted to explore the genome-wide m5C modification patterns in patients with cervical cancer, along with examination of their relationship with clinical features, such as tumor staging and prognosis. Second, investigation of the interactions between m5C modification and other RNA modifications (such as m6A and m1A) as well as their combined regulatory roles in cervical cancer cells may provide a new theoretical foundation for precision cancer therapy. Additionally, developing molecular tools to specifically regulate m5C methylation may offer therapeutic targets for cervical cancer treatment. In summary, our study scrutinized the expression profiles of 24 m5C regulators in CESE, uncovering their correlation with patient survival. By developing an m5C scoring system that integrates the assessment of individual m5C methylation patterns with the characteristics of corresponding TME cell infiltration, we aimed to facilitate personalized treatment plans and prognosis predictions for patients with CESE. Furthermore, m5C regulators or m5C-related genes may play a pivotal role in clinical responses to immunotherapy, ultimately enhancing survival outcomes for patients with CESE.
Supplemental Material
sj-pdf-1-imr-10.1177_03000605251328301 - Supplemental material for 5-Methylcytosine methylation predicts cervical cancer prognosis, shaping immune cell infiltration
Supplemental material, sj-pdf-1-imr-10.1177_03000605251328301 for 5-Methylcytosine methylation predicts cervical cancer prognosis, shaping immune cell infiltration by Luyang Su, Ren Xu, Yanan Ren, Shixia Zhao, Liyun Song, Cuiqiao Meng, Weilan Liu, Xuan Zhou and Zeqing Du in Journal of International Medical Research
Supplemental Material
sj-zip-2-imr-10.1177_03000605251328301 - Supplemental material for 5-Methylcytosine methylation predicts cervical cancer prognosis, shaping immune cell infiltration
Supplemental material, sj-zip-2-imr-10.1177_03000605251328301 for 5-Methylcytosine methylation predicts cervical cancer prognosis, shaping immune cell infiltration by Luyang Su, Ren Xu, Yanan Ren, Shixia Zhao, Liyun Song, Cuiqiao Meng, Weilan Liu, Xuan Zhou and Zeqing Du in Journal of International Medical Research
Footnotes
Acknowledgments
The authors acknowledge TCGA database and GEO database for providing the platform for uploading the meaningful datasets.
Authors’ contributions
Luyang Su and Zeqing Du conceived and designed the study. Ren Xu, Yanan Ren, and Shixia Zhao performed material preparation, data collection, and analysis. Liyun Song, Cuiqiao Meng, and Weilan Liu provided critical reagents and resources. Xuan Zhou conducted data curation and validation. Zeqing Du wrote the initial manuscript draft, and all authors contributed to manuscript revision. All authors have read and approved the final submitted version.
Availability of data and material
The datasets generated or analyzed during the current study are available in the GEO database (https://www.ncbi.nlm.nih.gov/geo/) and the TCGA database (![]()
Consent for publication
Not applicable.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Ethics approval and consent to participate
Ethical review and approval was not required for the study.
Funding
The authors report that there is no funding associated with the work featured in this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.