
Abstract
Objective
This study aimed to investigate cell-type-specific RNA methylation patterns in primary breast cancers and metastatic lymph nodes and explore their roles in cancer progression and immune regulation.
Methods
Using single-cell RNA sequencing data from five breast cancer patients (primary cancers and matched metastatic lymph nodes), we calculated RNA methylation scores per cell using the AUCell method. Cell types were annotated using Seurat. Differentially expressed gene analysis, pseudotime trajectory assessment, copy number variation analysis, and cell–cell communication analyses were performed between high- and low-methylation groups. Validation was conducted using bulk RNA sequencing data from The Cancer Genome Atlas.
Results
Metastatic lymph nodes showed significantly lower RNA methylation scores than primary cancers, particularly in T cells and B cells. Low methylation was correlated with enhanced cell differentiation, increased malignancy in epithelial cells, and reduced immune cell communication. Monocytes exhibited opposite trends. Differentially expressed genes from methylation groups stratified The Cancer Genome Atlas patients into two clusters with distinct survival and immune infiltration profiles.
Conclusions
RNA methylation is suppressed during breast cancer metastasis, affecting cell differentiation, immune communication, and patient outcomes.
Introduction
Breast cancer is a leading cause of mortality in females worldwide. 1 Apart from surgical resection or radiotherapy/chemotherapy, endocrine and targeted treatments remain limited. 2 Moreover, due to the heterogeneity and phenotypic plasticity of breast cancer, precision medicine continues to pose challenges. The identification of key pathways and driver genes involved in malignant metastasis provides opportunities for developing new treatments.3–5
It is well documented that cancer cells in metastatic sites exhibit diverse molecular and phenotypic properties compared with cells in primary cancers (PCs).6–7 Moreover, only a small fraction of cancer cells that leave PCs succeed in forming macroscopic metastases. 8 During malignant metastasis, RNA methylation is critical for the epigenetic and metabolic reprogramming of cancer cells as these cells adapt to new environments and evade immune surveillance.9–13 Identifying the key roles of RNA methylation in primary and metastatic tumor cells may offer comprehensive and unbiased measurements to identify patients who are more prone to metastasis or to guide the development of therapeutic strategies aimed at preventing metastasis.
The advent and rapid development of multiple single-cell technologies have provided opportunities to investigate cell-type-specific mechanisms of cancer metastasis at unprecedented resolution.14–16 Examination of the complex regulation of different cell ratios and cell–cell communications may assist in identifying aberrant cell types that could be therapeutically targeted.17–18 Identification of oncogenic or immunosuppressive signaling might also provide clues for restoring immune homeostasis.19–20 Likewise, single-cell technologies should be extensively used in studies of RNA methylation to help formulate more precise hypotheses for the identification of potential therapeutic interventions for breast cancer.
Currently, the integrative investigation of RNA methylation using single-cell transcriptomics in breast cancer remains limited. In light of the abovementioned findings, we attempted to provide a new approach to examine the role of RNA methylation in breast cancer metastasis. 21 Herein, we calculated RNA methylation scores for each cell and assessed score variations across samples and groups to validate cell-type-specific methylation patterns in breast cancer (Figure S1). The investigation of RNA methylation at the single-cell level may provide novel insights into the initiation of metastasis and the development of therapeutic interventions.
Materials and methods
Data preparation
The single-cell RNA sequencing (scRNA-seq) data of the PCs and two matched lymph nodes (LNs) from five breast cancer patients are publicly available in the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE180286). 22
The bulk RNA-seq data of breast cancer are publicly available in The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/repository).
The 50 hallmark pathways and RNA methylation-related genes are publicly available in the Gene Set Enrichment Analysis (GSEA) database (http://www.gsea-msigdb.org/gsea/downloads.jsp, v7.5.1). 23 All these genes have been repeatedly validated in the literature.
RNA methylation scores: calculation and comparison
Using the pipeline of the ‘Seurat’ (Version 4.3) R package 24 and established cell markers, we identified nine cell types. We calculated the cell ratios of PCs and metastatic LNs. We used the “AUCell” method 25 to estimate RNA methylation scores for every cell and compared the scores across all samples and cell types as well as between PCs and metastatic LNs.
Because RNA methylation scores were reduced in metastatic LNs, we additionally compared the scores for each cell type between PCs and metastatic LNs. All cells were stratified into high- and low-methylation groups based on their cell-type-specific median methylation scores. Differentially expressed genes (DEGs) between high- and low-methylation groups (|log2FC| > 0.5) were identified for each cell type. Furthermore, to clarify the underlying implications of these differences, the area under the curve (AUC) scores of the 50 hallmark pathways were compared between the two groups in PCs and metastatic LNs.
RNA methylation in epithelial cells
To understand the functions of RNA methylation in epithelial cells, we conducted pseudotime analysis using the ‘monocle’ (version 2.26) R package. We compared cell development and differentiation between the high- and low-methylation groups.
Based on the observed differences in cell development and differentiation, we used the ‘SCEVAN’ (version 1.0.1) R package, which calculates the copy number variation (CNV), to investigate the differences in malignancy between the two groups.
RNA methylation in monocytes
Because metastatic LNs are characterized by higher RNA methylation levels, we used the ‘monocle’ R package to compare differentiation and cell development. In addition, considering the differences in differentiation states between the two groups, we performed Gene Ontology (GO) enrichment analysis to investigate the difference between state 1 and state 4.
RNA methylation in B cells
When comparing metastatic LNs and B cells, the high-methylation group showed lower RNA methylation scores, and the low-methylation group exhibited the greatest number of DEGs. We therefore used the ‘monocle’ R package to calculate and compare cell development and differentiation between the high- and low-methylation groups. We examined genes with changing expression over pseudotime and classified them into clusters based on their expression patterns. Based on the differences in differentiation states between the two groups, we performed GO enrichment analysis to investigate the differences between state 1 and 7.
Cell communication differences
RNA methylation may influence cell–cell communications. Therefore, we used the ‘CellChat’ (version 1.4) R package 26 to calculate cell–cell communications and assess differences between the high- and low-methylation groups. Comparisons were performed for the number and strength of interactions between PCs and metastatic LNs as well as between high- and low-methylation groups.
Patient stratification based on DEGs
To explore the clinical relevance of RNA methylation and the results from single-cell data, we stratified TCGA patients into two clusters based on cell-type-specific DEGs between high- and low-methylation groups using the ‘NMF’ (version 0.26) R package. 27 We then used the ‘survival’ (version 3.5-3) R package to compare overall survival (OS) between the two clusters.
To investigate the underlying characteristics of the two clusters, we used the ‘clusterProfiler’ (version 4.6.2) R package to perform GO pathway enrichment analysis for clusters C1 and C2. Because immune-related pathways were enriched in C2, we calculated immune cell and pathway scores using the single-sample GSEA (ssGSEA) method and compared the scores between clusters C1 and C2.
Statistical analysis
All statistical analyses were conducted using R software (version 4.1.3). The Wilcoxon test was used to compare differences between the two groups. In all statistical analyses, P < 0.05 was considered statistically significant. Default parameters were used for all functions unless otherwise specified.
Results
Cell-type annotation and RNA methylation score comparison
Using the cell markers shown in Figure S2, we annotated 30 clusters into 9 cell types. The uniform manifold approximation and projection (UMAP) in Figure 1(a) illustrates the cell characteristics in two dimensions. As shown in Figure 1(b), PCs and metastatic LNs had different cell ratios, with a noticeable decrease in T cells and B cells and increase in epithelial cells and fibroblasts. The 74 well-documented RNA methylation-related genes are listed in Table S1. These genes were expressed at scaled levels and ratios in PCs and metastatic LNs (Figure S3). Comparisons of RNA methylation scores between PCs and metastatic LNs are shown in Figure 1(c). Metastatic LNs exhibited significantly lower methylation scores than PCs (Figure 1(d)). Figure 1(e) shows RNA methylation scores across all cell types.

Nine cell types were annotated, and RNA methylation scores were compared. (a) The UMAP showed the cell characteristics in two dimensions. (b) The cell ratios in PCs and metastatic LNs. (c) The UMAP of the RNA methylation scores in PCs and metastatic LNs. (d) RNA methylation scores were significantly lower in metastatic LNs than in PCs (***: P < 0.001) and (e) RNA methylation scores in all cell types. UMAP: uniform manifold approximation and projection; PCs: primary cancers; LNs: lymph nodes.
We categorized all cells into high- and low-methylation groups based on their median RNA methylation scores, and the DEGs between the two groups are listed in Table S2. As shown in Figure S4A, PCs and metastatic LNs exhibited significantly different RNA methylation scores. Monocytes had significantly higher RNA methylation scores than PCs, whereas the RNA methylation scores of other cell types, except dendritic cells, were significantly lower in metastatic LNs. Figure S4B shows the AUC scores for the 50 hallmark pathways in PCs and metastatic LNs stratified by high and low methylation. Compared with PCs, AUC scores for almost all pathways were lower in metastatic LNs. In PCs, the low-methylation group exhibited higher score for inflammation-related pathways and lower scores for tumor proliferation-related pathways.
RNA methylation related to differentiation and malignancy in epithelial cells
Figure 2(a) and (b) shows the potential pseudotime or states of all epithelial cells in the high- and low-RNA methylation groups. Longer pseudotime may indicate higher differential states. The high-methylation group contained more cells in state 1 and fewer cells in state 3 compared with the low-methylation group (Figure 2(c)). Similarly, the high-methylation group exhibited lower overall pseudotime (Figure 2(d)).
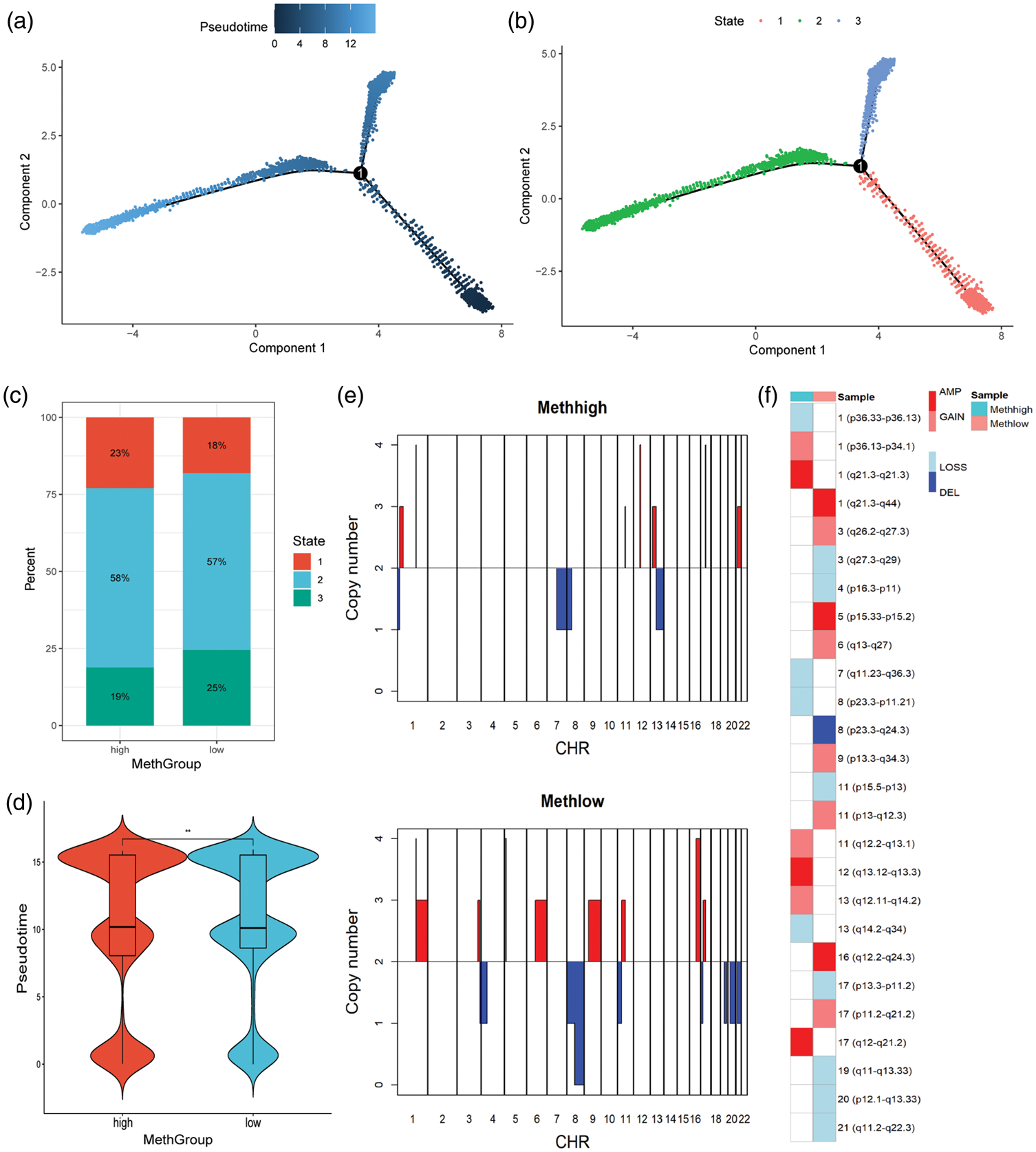
High methylation indicated less differentiation and lower malignancy in epithelial cells. (a–b) The potential pseudotime (a) or states (b) of all epithelial cells in high- and low-RNA methylation groups. (c) The high-methylation group contained more cells in state 1 and fewer cells in state 3 compared with the low-methylation group. (d) The high-methylation group exhibited lower pseudotime (***: P < 0.01). (e) Differing copy number profiles between high- and low-methylation groups and (f) Differing copy number variations between high- and low-methylation groups.
Figure 2(e) shows the differing copy number profiles, and Figure 2(f) shows the CNVs between high- and low-methylation groups. The low-methylation group appeared to have more CNVs.
Differentiation of monocytes indicated by RNA methylation
The pseudotime or states of monocytes in the high- and low-RNA methylation groups are shown in Figure S5A and S5B, respectively. The high-methylation group exhibited longer pseudotime than the low-methylation group (Figure S5C). Additionally, the high-methylation group contained more cells in state 4 and fewer cells in state 1 compared with the low-methylation group (Figure S5D). GO enrichment analysis of the DEGs between state 1 and state 4 indicated differences in chemotaxis function (Figure S5E).
Association of B cell differentiation with RNA methylation
The pseudotime of all B cells in the high- and low-RNA methylation groups is shown in Figure 3(a) and (b). The high-methylation group exhibited shorter pseudotime (Figure 3(c)). Additionally, the high-methylation group contained fewer cells in states 6 and 7 and more cells in state 1 compared with the low-methylation group (Figure 3(d)).

High methylation indicated less differentiation in B cells. (a–b) The potential pseudotime (a) or states (b) of all B cells in high- and low-RNA methylation groups. (c) The high-methylation group exhibited shorter pseudotime. (d) The high-methylation group contained fewer cells in states 6 and 7 and more cells in state 1 compared with the low-methylation group. (e) Expression profiles of the methylation genes changed significantly over the pseudotime and (f) GO enrichment analysis of the DEGs in states 1, 6, and 7 indicated the differences in immune function. DEGs: differentially expressed genes; GO: Gene Ontology.
Considering the significant pseudotime variation among B cells, Figure 3(e) illustrates the expression profiles of the methylation-related genes, which showed substantial changes across pseudotime, predominantly decreasing over time. GO enrichment analysis of the DEGs in states 1, 6, and 7 indicated differences in immune function, including receptor for advanced glycation end products (RAGE) receptor binding (Figure 3(f)).
Relation of cell–cell communications with RNA methylation
Figure 4(a) and (b) illustrates cell–cell communications in PCs and metastatic LNs, respectively. Figure S6A and S6B shows the number and strength of inferred interactions between PCs and metastatic LNs. The interaction strength in metastatic LNs decreased compared with that in PCs, suggesting potential immune escape. Figure S6C displays the specific interactions in PCs and metastatic LNs. Metastatic LNs lost COMPLEMENT and major histocompatibility complex class I (MHC-I) interactions, indicating impaired immune function.

High methylation indicated less cell communication. (a–b) Cell–cell communications of PCs (a) and metastatic LNs (b). (c–d) Number (c) and strength (d) of inferred interactions between high- and low-methylation groups. (e) Specific interactions in high- and low-methylation groups and (f) overall signaling patterns of high- and low-methylation groups. PCs: primary cancers; LNs: lymph nodes.
Figure 4(c) and (d) shows the number and strength of inferred interactions between high- and low-methylation groups, and Figure 4(e) displays the specific interactions in these groups. MHC-I interactions in the high-methylation group suggested better differentiation of innate immune cells, such as monocytes, consistent with Figure S5C and S5D. Figure 4(f) shows the overall signaling patterns of high- and low-methylation groups. Signaling pathways including MHC-I, SELPLG, VEGF, CDH1, COMPLEMENT, and MPZ differed between the two groups, indicating that these pathways may be influenced by RNA methylation.
Classification of patients via DEGs of RNA methylation
To explore the clinical relevance of cell-type-specific DEGs from high- and low-methylation groups, we stratified TCGA patients into two clusters. Prognosis-related genes could efficiently classify patients into these clusters (Figure 5(a)). OS was better in cluster C2 than in cluster C1 (Figure 5(b)).

Classification of patients based on DEGs from different methylation groups. (a) Prognostic cell-type-specific DEGs efficiently classified patients into two clusters. (b) OS of cluster C2 was better than cluster C1. (c) GSEA of the DEGs between clusters indicated that C1 was enriched in arachidonic acid monooxygenase activity. (d) C2 was mainly enriched in immune-related functions and (e) ssGSEA revealed higher immune infiltration of cluster C2. DEGs: differentially expressed genes; OS: overall survival; GSEA: gene set enrichment analysis; ssGSEA: single-sample GSEA.
GSEA of the DEGs between clusters indicated that C1 was enriched in arachidonic acid monooxygenase activity (Figure 5(c)), whereas C2 was primarily enriched in immune-related functions (Figure 5(d)). ssGSEA also showed higher immune infiltration in cluster C2 (Figure 5(e)).
Discussion
Despite decades of research, many aspects of the mechanisms and functions of RNA methylation in tumors remain unclear. 28 Traditional methods have established that RNA methylation plays key roles in the initiation and progression of cancer.29–30 However, these approaches lack consistency and specificity across diverse contexts and have not translated into effective treatments due to the absence of an integrative and comprehensive understanding. Novel technologies, such as next-generation sequencing and single-cell multi-omics, provide new insights for maintaining and restoring cellular homeostasis. 31 In our study, we depicted the cell-type-specific landscape of RNA methylation in PCs and metastatic LNs as well as cell–cell interactions, reflecting phenotypic variability. The decrease in T cell and B cell ratios indicated deficiencies in adaptive immunity within metastatic LNs. Furthermore, RNA methylation scores were decreased in T cells and B cells in metastatic LNs, which may be associated with the reduction in cell ratios.
Mechanistically, RNA methylation may be associated with cancer metastasis in breast cancer, 32 which is supported by our findings showing that the main differences between high- and low-methylation groups involved inflammation, proliferation, and immune-related pathways. Moreover, RNA methylation has been linked to cell differentiation, 33 which is consistent with our observation that different methylation groups exhibited distinct cell differentiation states. Epithelial cells in the low-methylation group showed significantly higher differentiation states compared with those in the high-methylation group. Additionally, the low-methylation group appeared to have more CNVs than the high-methylation group. These observations suggest a negative correlation between RNA methylation and breast cancer malignancy, in agreement with previous studies.
Our findings indicated that RNA methylation in T cells from metastatic LNs was downregulated, which may promote immune escape through multiple mechanisms. First, previous studies have shown that methylation is essential for maintaining T cell homeostasis and function; for example, loss of METTL3 impairs T cell responses to interleukin-7 (IL-7) signaling, leading to exhaustion. 34 Second, RNA methylation also finely regulates the differentiation of specific T cell subsets, such as T follicular helper cells, 35 and its downregulation may broadly disrupt the coordination of adaptive immune responses, as observed similarly in B cells. 36 Therefore, the hypomethylated T cells we observed were likely in a functionally impaired state. More importantly, these functionally deficient T cells, combined with the loss of MHC-I signaling in tumor cells, create a “double-hit” scenario for immune escape. 37 We propose that RNA methylation reprogramming is a key factor in shaping the immunosuppressive tumor microenvironment, 29 and its regulatory mechanisms may serve as novel targets for improving cancer immunotherapy.
Interestingly, monocytes exhibited RNA methylation patterns that differed from those of other cell types. The low-methylation group showed significantly lower differentiation states compared with the high-methylation group, which was the opposite to what was observed in other cell types, such as adaptive immune and epithelial cells. GO enrichment analysis of the DEGs in states 1 and 4 indicated differences in antigen processing, cell clearance, and chemotaxis functions. In contrast, B cells displayed RNA methylation patterns consistent with those of epithelial cells. The low-methylation group exhibited higher differentiation states compared with the high-methylation group, suggesting a relatively strong influence of RNA methylation in B cells, as also reflected by the highest ratios of cell-type-specific DEGs between high- and low-methylation groups shown in Table S2. Nearly half of the RNA methylation-related genes changed significantly over pseudotime, with most showing decreased expression. GO enrichment analysis of the DEGs in states 1, 6, and 7 indicated differences in B cell differentiation. Notably, the RAGE receptor binding ranked first, highlighting the importance of RAGE in B cell differentiation and its close association with RNA methylation.
Distinct cell–cell communication patterns exist between PCs and metastatic LNs. As reported in previous studies, specific signaling pathways present in PCs, such as COMPLEMENT and MHC-I, were lost in metastatic LNs.37,38 Macrophage migration inhibitory factor (MIF) and CD34, specifically observed in metastatic LNs, may reflect immunosuppressive characteristics and stemness.39–40 Similarly, the distinct signaling patterns observed in the low-methylation group, including integrin subunit beta 2 (ITGB2) and cadherin 1 (CDH1), indicate increased malignancy, consistent with previous findings. 41 Moreover, the loss of MHC-I and selectin P ligand (SELPLG) signaling in the low-methylation group suggests reduced immune function.
Non-negative matrix factorization (NMF) is an effective method for decomposing a matrix into two matrices. 34 Using predictive cell-type-specific DEGs, all samples in TCGA dataset were divided into two clusters. GSEA indicated that cluster C2 was more enriched in immune infiltration, which was also confirmed by ssGSEA. Integrated analysis with single-cell data suggested that RNA methylation influenced adaptive immune cell differentiation and, consequently, immune infiltration and patient outcomes. Li HB et al. demonstrated that knockout of METTL3 (methyltransferase 3) blocks T cell proliferation and differentiation by targeting IL-7/STAT5/SOCS pathways. 36 Deficiency of the m6A “writer” METTL14 was also reported to impair IL-7-induced B cell proliferation and maturation in mice. 42 METTL3 knockouts reduce translation of SPRED2 (sprouty-related EVH1 domain-containing 2) via YTHDF1 (YTH N6-methyladenosine RNA binding protein 1), thereby activating NF-kB (nuclear factor kappa B subunit 1) and STAT3 (signal transducer and activator of transcription 3) through MAPK1 (mitogen-activated protein kinase 1), which enhances tumor growth and metastasis. 24 These findings support our observation that inflammation-related pathways were elevated in the low-RNA methylation group.
Several limitations of our study should be acknowledged. First, we calculated an overall methylation score rather than type-specific methylation scores. Second, we did not provide validation data related to immunotherapy, which is of considerable importance. Currently, no suitable cohorts exist that include both breast cancer immunotherapy information and transcriptomic data. Therefore, we performed an alternative analysis using available data. In TCGA cohort, we found that a distinct patient cluster, defined by cell-type-specific DEGs from high- and low-methylation groups, was significantly associated with an immunosuppressive microenvironment, characterized by reduced CD8+ T cells and increased M2 macrophages. This “cold tumor” phenotype is generally predictive of a poorer response to immunotherapy. These findings indirectly support the hypothesis that the methylation score may be linked to immunotherapy outcomes. In addition, the sample size in our study was relatively small, and selection bias may be present. Generally, single-cell sequencing detects RNA expression but does not directly measure RNA methylation levels. Therefore, discrepancies may exist between “methylation scores based on gene expression” and “actual methylation levels.” The upstream mechanisms regulating methylation were also not analyzed. Nevertheless, our study demonstrated that RNA methylation plays a crucial role in regulating breast cancer cell differentiation and communication. These findings advance the understanding of RNA methylation in immune escape and immunotherapy. Future studies using larger cohorts,43–44 additional omics approaches (e.g. single-cell m6A sequencing, genome-wide analyses),45–48 or experimental investigations of upstream mechanisms in animal models, such as mice and primates,49–51 are warranted.
Conclusion
In summary, RNA methylation is suppressed during breast cancer metastasis. Metastatic LNs exhibited low RNA methylation in adaptive immune cells and high methylation in innate immune cells. Moreover, RNA methylation influences cell differentiation and intercellular communications. Classification of bulk RNA-seq data using cell-type-specific DEGs further highlighted the role of RNA methylation in adaptive immunity. This study provides new insight into the role of RNA methylation in breast cancer progression.
Supplemental Material
sj-jpg-1-imr-10.1177_03000605251405638 - Supplemental material for Single-cell analysis reveal cell-type-specific RNA methylation patterns in breast cancer
Supplemental material, sj-jpg-1-imr-10.1177_03000605251405638 for Single-cell analysis reveal cell-type-specific RNA methylation patterns in breast cancer by Shengtao Huang, Yichen Lin, Xiaobing Liu, Kun Zhang, Qiuju Han, Ruilei Liu and Yong Zhuang in Journal of International Medical Research
Supplemental Material
sj-pdf-2-imr-10.1177_03000605251405638 - Supplemental material for Single-cell analysis reveal cell-type-specific RNA methylation patterns in breast cancer
Supplemental material, sj-pdf-2-imr-10.1177_03000605251405638 for Single-cell analysis reveal cell-type-specific RNA methylation patterns in breast cancer by Shengtao Huang, Yichen Lin, Xiaobing Liu, Kun Zhang, Qiuju Han, Ruilei Liu and Yong Zhuang in Journal of International Medical Research
Supplemental Material
sj-pdf-3-imr-10.1177_03000605251405638 - Supplemental material for Single-cell analysis reveal cell-type-specific RNA methylation patterns in breast cancer
Supplemental material, sj-pdf-3-imr-10.1177_03000605251405638 for Single-cell analysis reveal cell-type-specific RNA methylation patterns in breast cancer by Shengtao Huang, Yichen Lin, Xiaobing Liu, Kun Zhang, Qiuju Han, Ruilei Liu and Yong Zhuang in Journal of International Medical Research
Supplemental Material
sj-pdf-4-imr-10.1177_03000605251405638 - Supplemental material for Single-cell analysis reveal cell-type-specific RNA methylation patterns in breast cancer
Supplemental material, sj-pdf-4-imr-10.1177_03000605251405638 for Single-cell analysis reveal cell-type-specific RNA methylation patterns in breast cancer by Shengtao Huang, Yichen Lin, Xiaobing Liu, Kun Zhang, Qiuju Han, Ruilei Liu and Yong Zhuang in Journal of International Medical Research
Supplemental Material
sj-pdf-5-imr-10.1177_03000605251405638 - Supplemental material for Single-cell analysis reveal cell-type-specific RNA methylation patterns in breast cancer
Supplemental material, sj-pdf-5-imr-10.1177_03000605251405638 for Single-cell analysis reveal cell-type-specific RNA methylation patterns in breast cancer by Shengtao Huang, Yichen Lin, Xiaobing Liu, Kun Zhang, Qiuju Han, Ruilei Liu and Yong Zhuang in Journal of International Medical Research
Supplemental Material
sj-pdf-6-imr-10.1177_03000605251405638 - Supplemental material for Single-cell analysis reveal cell-type-specific RNA methylation patterns in breast cancer
Supplemental material, sj-pdf-6-imr-10.1177_03000605251405638 for Single-cell analysis reveal cell-type-specific RNA methylation patterns in breast cancer by Shengtao Huang, Yichen Lin, Xiaobing Liu, Kun Zhang, Qiuju Han, Ruilei Liu and Yong Zhuang in Journal of International Medical Research
Supplemental Material
sj-xlsx-7-imr-10.1177_03000605251405638 - Supplemental material for Single-cell analysis reveal cell-type-specific RNA methylation patterns in breast cancer
Supplemental material, sj-xlsx-7-imr-10.1177_03000605251405638 for Single-cell analysis reveal cell-type-specific RNA methylation patterns in breast cancer by Shengtao Huang, Yichen Lin, Xiaobing Liu, Kun Zhang, Qiuju Han, Ruilei Liu and Yong Zhuang in Journal of International Medical Research
Supplemental Material
sj-xlsx-8-imr-10.1177_03000605251405638 - Supplemental material for Single-cell analysis reveal cell-type-specific RNA methylation patterns in breast cancer
Supplemental material, sj-xlsx-8-imr-10.1177_03000605251405638 for Single-cell analysis reveal cell-type-specific RNA methylation patterns in breast cancer by Shengtao Huang, Yichen Lin, Xiaobing Liu, Kun Zhang, Qiuju Han, Ruilei Liu and Yong Zhuang in Journal of International Medical Research
Footnotes
Acknowledgments
Not applicable.
Author contributions
S.H. conceived the study, participated in data analysis, and drafted the manuscript.
R.L. and Y.Z. conceived the study and drafted the manuscript.
Y.L. participated in data analysis and manuscript revision.
X.L. and K.Z. participated in data analysis.
Q.H. participated in manuscript revision.
Data availability statement
The single-cell data are available in the GEO database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE180286). The bulk RNA-seq data are available in TCGA (![]() ) database.
) database.
Declaration of conflicting interests
The authors declare no conflicts of interest.
Institutional review board statement
Not applicable.
Informed consent statement
Not applicable.
Funding
This work was supported by the Science and Technology Innovation Foundation at the Fujian Provincial Maternal and Child Hospital of China (YCXY 23-08), the Natural Science Foundation of Fujian Province (2021J01214), the Startup Fund for Scientific Research, Fujian Medical University (2020QH1064), and the National Natural Science Foundation of China (82302377).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.