
Abstract
Inflammatory linear verrucous epidermal nevus (ILVEN) is a rare type of epidermal nevus characterized by an inflammatory appearance, intense pruritus, and a tendency to be resistant to treatment. We present here, the case of a 7-year-old boy who presented with a widespread inflammatory skin lesion on the right side of his body, with some of the eruption following Blaschko’s lines. He also had an associated contracture in the ipsilateral limb and short stature. Initially, we suspected this may be a rare case of CHILD syndrome in a boy. However, after further investigation, we confirmed the diagnosis as a severe form of ILVEN.
Background
Inflammatory linear verrucous epidermal nevus (ILVEN) a rare skin condition characterized by an inflamed verrucous plaque with severe pruritus and refractory to therapy, often occurs in early childhood.1,2 Another uncommon skin condition that presents with a unilateral red, scaly eruption following a Blaschko line pattern is CHILD syndrome which is characterized by congenital hemidysplasia, ichthyosiform erythroderma, and limb defects. 3 CHILD syndrome mimics not only the skin lesions, but also the histology of ILVEN. However, pronounced ipsilateral limb hypoplasia is pathognomonic for CHILD syndrome. 3 We describe here, an interesting case that presented with severe, unilateral skin lesions and ipsilateral limb abnormalities, which was ultimately diagnosed as ILVEN through gene sequencing.
Case report
A boy, aged between 6 and 8 years, presented to our department (Department of Dermatology, Tianjin Children's Hospital, Tianjin City, China) in August 2019, with a widespread rash affecting almost the entire right side of his body, while sparing his face. Erythematous papules and plaques were sharply demarcated along the midline of his abdomen and perineum (including the genitals), and appeared in a linear pattern following Blaschko's lines on the thorax and right limbs, including the palms and soles. The reddish plaques were covered with yellow waxy scales and partial excoriation was noted (Figure 1). The boy arrived in a wheelchair due to his inability to walk, caused by a flexion contracture of the right lower extremity. He also experienced constant, intolerable pruritus and hypersensitivity to environmental stimuli, such as cold and wind. His height was notably below average for children of his age and sex, although his intellectual development was within the average range. According to his parents, who were unrelated and in excellent health, the boy first showed the skin lesion at a young age. At that time, the erythema was localized and mild, and his extremities were fully functional with normal movement.
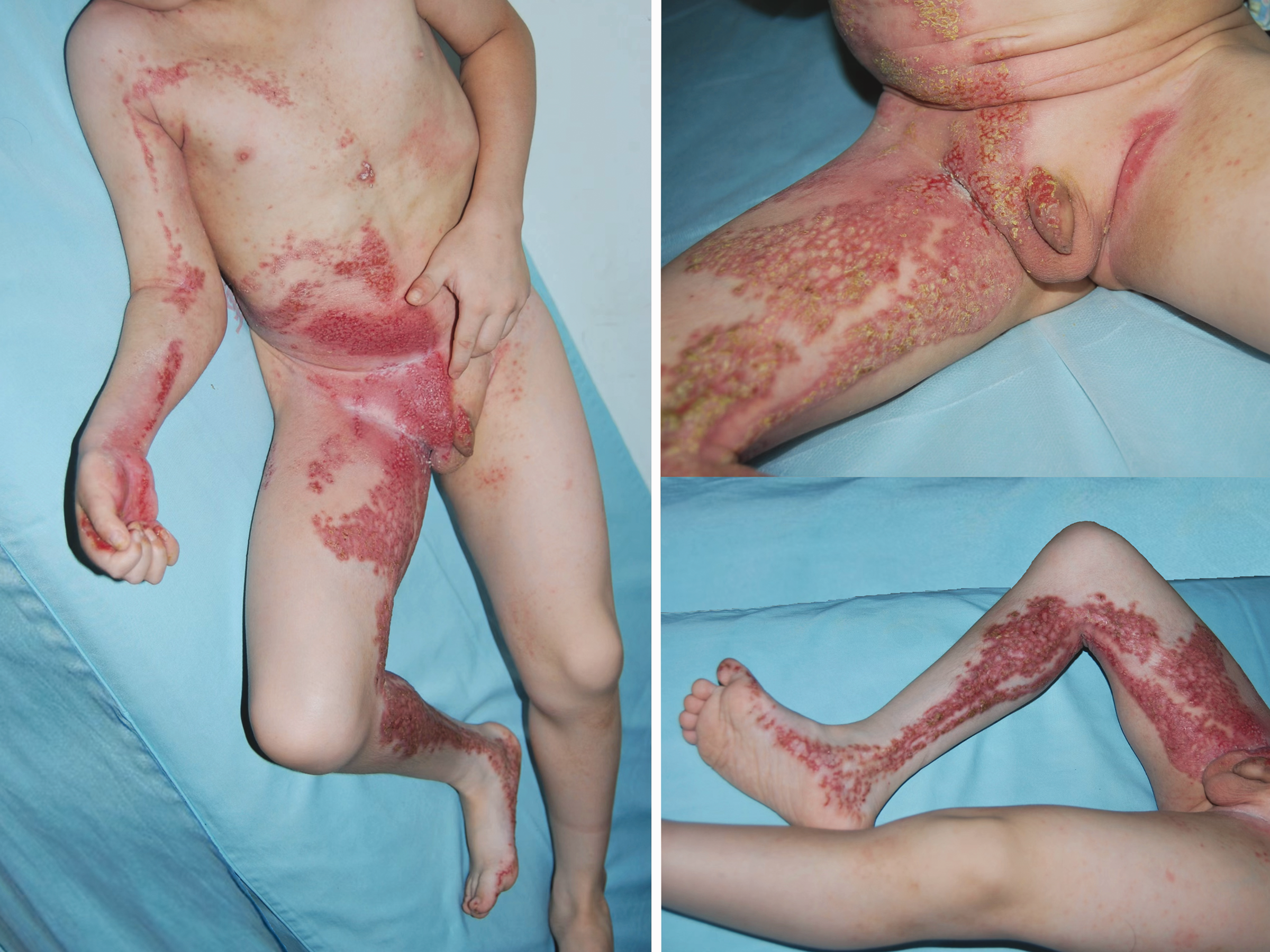
Reddish plaques covering the right half of the body, sharply demarcated along the midline and partly linear along the line of Blaschko.
Laboratory tests and abdominal ultrasonography (including liver, spleen, and kidneys) findings were normal or negative. A tissue sample for biopsy was taken from the child’s right thigh. Histological analysis showed the presence of acanthosis of the epidermis, accompanied by spotty areas of parakeratosis and elongated rete ridges, in association with bandlike lymphocytic and scattered histiocytic inflammation in the upper dermis. Foam cells were not observed (Figure 2). A spiral computed tomography (CT) plain scan plus 3 D reconstruction was taken to examine the child’s right extremities. The report indicated that all bones and joints were in normal condition, with no signs of epiphyseal stippling or hypoplasia in any of the bones that were examined.

In the acanthotic epidermis, spotty areas of parakeratosis were observed. In the upper dermis, bandlike lymphocytic and scattered histiocytic inflammation were noted. Haematoxylin and Eosin staining was applied, with the upper image captured at 10× magnification and the lower image at 20× magnification.
Blood and lesional skin tissue samples were sent for medical exome gene sequencing to investigate potential genetic diseases and as expected, we received negative reports. This, along with all the other clinical findings, led to a final diagnosis of ILVEN rather than CHILD syndrome. Following parents’ consent for treatment, we prescribed topical corticosteroids, topical calcineurin inhibitors, and calcipotriol in succession. However, due to inconsistent use, the improvement was limited and short-lived. At the five-year follow-up, the child was found to be in a stable condition, with no signs of further deterioration or improvement. The reporting of this study conforms to CARE guidelines. 4 The child’s parents provided consent for publication of his anonymised data.
Discussion
ILVEN is a rare skin disorder fist described and defined in 1971. 1 Even rarer than ILVEN, CHILD syndrome has a completely different epidemiological profile as the mutation in the X-linked dominant gene, NAD(P)-dependent steroid dehydrogenase-like (NSDHL), is lethal to male embryos. 3 However, in an extremely rare instance, an acquired postzygotic mutation or a gametic half-chromatid mutation in the NSDHL gene did result in a case of CHILD syndrome in a boy. 5 Otherwise, this condition is almost exclusively observed in girls.
The cause of ILVEN is unknown. Scientists have tried to identify the causative agent at the genetic level and have had some success. For example, a somatic heterozygous mutation in GJA1 6 and heterozygous missense variants in the CARD14 gene 2 have both been reported to contribute to some cases of ILVEN. These findings may prove useful in differentiating ILVEN from CHILD syndrome. However, these genes were not identified in our samples.
Distinguishing CHILD syndrome from ILVEN based solely on similar cutaneous signs is typically challenging. The intense pruritus seen in ILVEN and the ptychotropism (affinity for body folds) observed in CHILD syndrome 7 are characteristic of these two rare skin conditions, but neither is pathognomonic. Other common features of CHILD syndrome include skeletal abnormalities, although the extent and severity of involvement can vary.2,7 In our case, we confirmed that there was no underlying bone or joint pathology. To provide a rational explanation for the flexion contracture of our patient's right lower extremity, we considered the following: the chronic process of repeated erythema, oedema, erosion, scabbing, and self-repair likely resulted in histological changes, including fibrosis, which was similar to scarring. The proliferation of fibrous tissue in the trans-articular region resulted in a reduction in the range of joint movement.
On histopathology, verruciform xanthoma, (foamy, lipid-laden histiocytes in dermal papillae) may help distinguish CHILD syndrome from other rare skin conditions.2,7 However, other histological features are non-specific. Interestingly, when biopsies are not feasible, reflectance confocal microscopy (RCM) has been used to diagnose ILVEN. 8 RCM can show minimal hyperkeratosis, spongiosis, and distinctive signs of interface dermatitis. 8
Recent studies have attempted to differentiate ILVEN from other diseases through techniques such as immunohistochemical staining, pathophysiology analysis, and proteomics. While these approaches show promise, they are still emerging and not yet sufficient for a definitive diagnosis or treatment. Both of these rare conditions, ILVEN and CHILD syndrome, continue to require further research.