
Abstract
Objective
To investigate the cerebroprotective effects of leptin in vitro and in vivo via the Janus kinase-2 (JAK2)/transcription factor signal transducer and activators of transcription-3 (STAT3) pathway and leptin receptors (LEPR).
Methods
The study used the cellular oxygen-glucose deprivation (OGD) model in PC12 cells and the middle cerebral artery occlusion (MCAO) rat model of cerebral ischaemia–reperfusion injury (CIRI) to assess changes in gene expression and protein levels following leptin pretreatment. The methylated DNA immunoprecipitation (MeDIP) assay measured DNA methylation levels.
Results
The optimal leptin concentration for exerting neuroprotective effects against ischaemia–reperfusion injury in PC12 cells was 200 ng/ml in vitro, but excessive leptin diminished this effect. Leptin pretreatment in the MCAO rat model demonstrated a similar effect to previously reported leptin administration post-CIRI. In addition to regulating the expression of inflammation-related cytokines, Western blot analysis showed that leptin pretreatment upregulated BCL-2 and downregulated caspase 3 levels. The MeDIP analysis demonstrated that DNA methylation regulated LEPR gene expression in the MCAO rat model when leptin pretreatment was used.
Conclusion
Exogenous leptin might bind to extra-activated LEPR by reducing the methylation level of the LEPR gene promoter region, which leads to an increase in phosphorylated JAK2/STAT3 and apoptotic signalling pathways.
Introduction
Cerebrovascular disease (CVD) is a prevalent and substantial health concern; with it being the third leading cause of mortality after heart disease and cancer. 1 Approximately 2 million new cases of stroke are reported annually, of which 60% are ischaemic strokes. 2 Three-quarters of patients treated for stroke present with varying degrees of sequelae, resulting in the loss of self-care and work capacity, in addition to the substantial burden on family and society.1–3 Modern treatments have significantly improved the survival and prolonged the life of patients with CVD, but the existing treatments pose the risk of cerebral ischaemia–reperfusion injury (CIRI). 2 In CIRI, brain function fails to recover, accompanied by the development of more severe neurological dysfunction after blood flow restoration to the ischaemic area of brain tissue. CIRI is a severe neurological dysfunction that occurs during the restoration of blood supply to the ischaemic area. The underlying pathophysiological mechanisms are principally related to the formation of oxygen free radicals, calcium overload, neurotransmitter toxicity and endothelial cell damage.2,4–6
Leptin is an adipose-derived hormone secreted by the adipocytes that is encoded by the LEP gene. 7 Leptin expression is primarily modulated by neuroendocrine feedback mechanisms and energy metabolism. 8 Leptin plays a crucial role in the hypothalamic regulation of food intake and energy balance. 9 Furthermore, it participates in the modulation of immune responses, neuronal development and post-injury survival of neurons.7–9 Leptin is closely associated with the development of tissue ischaemia and ischaemia–reperfusion injury. 7 It is central to energy metabolism and damage repair in the liver and cardiomyocytes.10,11 Moreover, leptin can reduce tissue reperfusion injury after ischaemia. 9 A previous study showed that exogenous leptin administered into the lateral ventricle of mice binds to leptin receptors (LEPR) in the central nervous system, specifically in regions such as the hypothalamus, hippocampus and cortex. 12 This binding activates Janus kinase-2 (JAK2) on the neuronal cell membrane, followed by its phosphorylation. 12 Subsequently, it activates transcription factor signal transducer and activators of transcription-3 (STAT3). 12 Phosphorylated STAT3 (p-STAT3) is implicated in the regulation of target gene expression.13–15 P-STAT3 exerts a trophic and protective effect on the nerves.16–18 However, local aggregation of leptin also leads to cell apoptosis. 19 The optimal concentration of leptin to exert neuroprotective effects remains to be established.
This current study aimed to explore whether more convenient administration routes, such as intraperitoneal injection, can exert comparable neuroprotective effects of leptin in brain tissue. In addition, the reason for the change in LEPR levels in brain tissue after leptin pretreatment and their specific role in activating the JAK2/STAT3 signalling pathway remains to be investigated. Therefore, this current study also investigated the control of LEPR levels and the role of epigenetics.
Materials and methods
Constructing the cellular oxygen-glucose deprivation (OGD) cell model
Adrenal phaeochromocytoma (PC12) cells (Cell Bank, Chinese Academy of Science, Shanghai, China) in the experimental group were pretreated with different leptin concentrations (0 ng/ml, 50 ng/ml, 100 ng/ml, 200 ng/ml or 300 ng/ml) for 30 min. The cells were cultured in sugar-free medium (GIBCO® Cell Culture, Carlsbad, CA, USA) and incubated in a three-gas incubator (Anoxomat® III Anaerobic Culture System; Mart Microbiology BV, Drachten, The Netherlands) consisting of 0% O2, 5% CO2 and 95% N2 at 37°C for l h. Then glucose-containing medium was added and the cells were incubated in a normal oxygen-saturated CO2 incubator (Anoxomat® III Anaerobic Culture System; Mart Microbiology BV) for up to 24 h. The control group of cells was exposed to the same leptin concentrations but no other treatments were administered.
High-sensitivity Cell Counting Kit-8 (CCK-8) cytotoxicity assay
The viability of the OGD cell model in response to different leptin concentrations and reperfusion times was measured using the KGI Bio high-sensitivity CCK-8 assay kit according to the manufacturer’s instructions (KGA317; Keygen, Wuxi, Jiangsu, China).
Middle cerebral artery occlusion (MCAO) rat model
Sprague-Dawley rats (n = 30) weighing approximately 250–280 g and aged 8–9 weeks were sourced from Viton Lever Ltd. (Beijing, China). The mean weight of the groups of Sprague-Dawley rats before and after the pretreatment procedure are shown in Table 1. Prior to the experiment, all rats were individually housed for 7 days at the Animal Experimentation Centre (Wuhan University, Wuhan, China). They were kept under a 12-h light/12-h dark cycle with free access to food and water to alleviate mental stress. This animal study was conducted in accordance with the Declaration of Helsinki and the Guidelines for the Treatment of Animals in China. The experimental protocol was approved by the Ethics Committee of Zhongnan Hospital of Wuhan University (no. ZN2021196).
The mean weight of the groups of Sprague-Dawley rats before and after the pretreatment procedure.
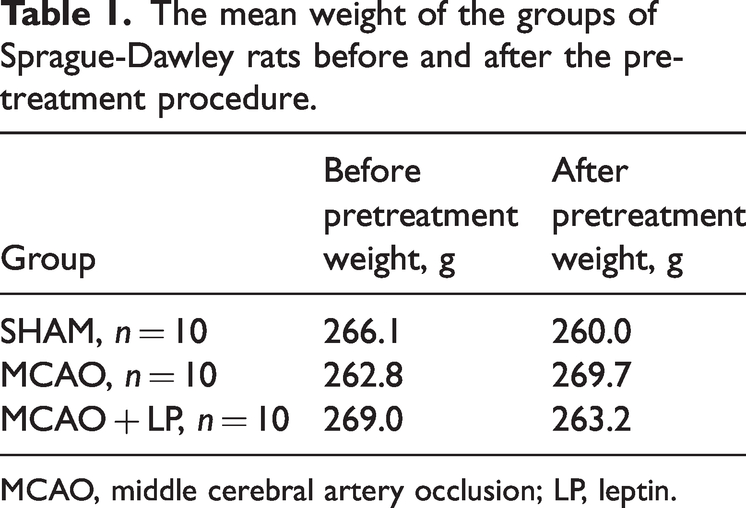
MCAO, middle cerebral artery occlusion; LP, leptin.
The MCAO rat model was constructed using the modified Longa wire embolization method.20,21 Rats were anaesthetized via intraperitoneal injection of 7% chloral hydrate (0.3 ml/100 g) and positioned supine on the operating table. Following neck disinfection, a 2-cm incision was made in the middle of the neck to expose the common carotid artery. The distal end of the artery was ligated 0.5 cm from its bifurcation and the proximal end was loosely ligated and clamped with a vascular clip. A small incision was made between the ligatures and a suture was inserted towards the internal carotid artery until it reached the bifurcation of the middle cerebral artery (approximately 18 mm insertion length). Post-surgery, the rats were placed on a thermostatic blanket to maintain an intra-anal temperature of 37 ± 0.5°C. The suture was removed after 1 h of ischaemia and rats were reperfused for an additional 24 h. Only rats with modified Longa scores ranging from 1 to 3 were included in the study. Animals displaying signs of subarachnoid haemorrhage post-surgery were excluded from the experimental group irrespective of any ischaemic symptoms. A total of eight rats were excluded from the study.
The rats were divided into three groups: (i) sham operation group (SHAM, n = 10) in which the common carotid, external carotid and internal carotid arteries were isolated without ligation or embolization; (ii) cerebral ischaemia–reperfusion model control group (MCAO, n = 10) in which saline (20 μl, 0.9%) was injected intraperitoneally daily at the same time each day for 1 week prior to the MCAO procedure; (iii) MCAO preoperative leptin preconditioning group (MCAO + LP, n = 10) in which recombinant mouse leptin (10 μl) was injected intraperitoneally at a dose of 200 ng/ml daily at the same time each day for 1 week prior to the MCAO procedure.
After 1 h of ischaemia and 24 h of reperfusion, the rat brains were removed and placed on ice. Then, 1-mm coronal sections were sliced and incubated in 2% 2,3,5-triphenyltetrazolium chloride (TTC) at 37°C for 30 min. Fixed brain sections were then photographed and analysed using Image J software (version 1.8.0, National Institutes of Health, Bethesda, MD, USA) to calculate the infarcted area. Brain tissues from the infarcted area were stained with haematoxylin and eosin (H&E). Slides were processed for dephosphorylation and rehydration before staining with H&E. Images were captured using an inverted phase contrast microscope (magnification: 100×, 400×; Leica DMi1; Leica, Solms, Germany).
Detecting protein levels and changes in DNA and RNA levels
Total protein was extracted from OGD cell lines and rat brain tissues using RIPA Lysis and Extraction Buffer (Beyotime Biotechnology, Shanghai, China) and quantified using a BCA protein assay reagent kit (Beyotime Biotechnology). The proteins were separated by 10% sodium dodecyl sulphate–polyacrylamide gel electrophoresis at 110 V for 70 min. The proteins were then transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA) using electroblot apparatus at 350 mA for 50 min (Bio-Rad, Hercules, CA, USA). The membranes were incubated in blocking solution consisting of Tris-buffered saline-Tween 20 (TBST; pH 7.5; 20 mmol/l Tris–HCl, 150 mmol/l sodium chloride, 0.1% Tween-20) containing 5% nonfat milk at room temperature for 1 h. The membranes were then incubated with primary rabbit antirat antibodies for JAK2, STAT3, p-JAK3, p-STAT3, IL-6, IL-10, caspase 3, BCL-2 and GAPDH (dilution 1:1000; Beyotime Biotechnology) at 4°C overnight. The membranes were then washed with TBST (pH 7.5) three times and incubated with horseradish peroxidase-conjugated goat antirabbit secondary antibody (dilution 1:4000; Beyotime Biotechnology) for 1 h at room temperature. Then the membranes were then washed with TBST (pH 7.5) three times. The membranes were developed using an enhanced chemiluminescence reagent kit (Bio-Rad) and exposed to X-radiography film. Immunoblots were scanned using a densitometer (Bio-Rad) and analysed using Quantity One software, version 4.6.2 (Bio-Rad).
Total DNA was extracted from OGD cell lines and rat brain tissues using FastPure® Cell/Tissue DNA Isolation Mini Kit (Vazyme, Nanjing, Jiangsu, China) according to the manufacturer’s instructions. Total RNA was extracted from OGD cell lines and rat brain tissues using TRIzol reagent (Takara Bio, Kyoto, Japan) according to the manufacturer’s instructions. Total RNA was reverse transcribed using a HiScript III 1st Strand cDNA Synthesis Kit (Vazyme) according to the manufacturer’s instructions. The resulting cDNA was used for real-time polymerase chain reaction (PCR) using ChamQ Universal SYBR qPCR Master Mix (Vazyme) in triplicates using a Rotor-Gene Q Real Time System (Qiagen Inc., Shanghai, China). The primer sequences for the real-time quantitative PCR analysis are shown in Table 2. All primers were synthesized by Sangon Biotech (Shanghai, China). The cycling programme involved preliminary denaturation at 95°C for 30 s, followed by 45 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s and elongation at 72°C for 45 s, followed by a final elongation step at 72°C for 5 min. The relative mRNA levels were compared with those of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and calculated using the 2-△CT method. The CT value used for these calculations was the mean of the triplicate for each reaction.
Primer sequences for real-time quantitative polymerase chain reaction analyses.
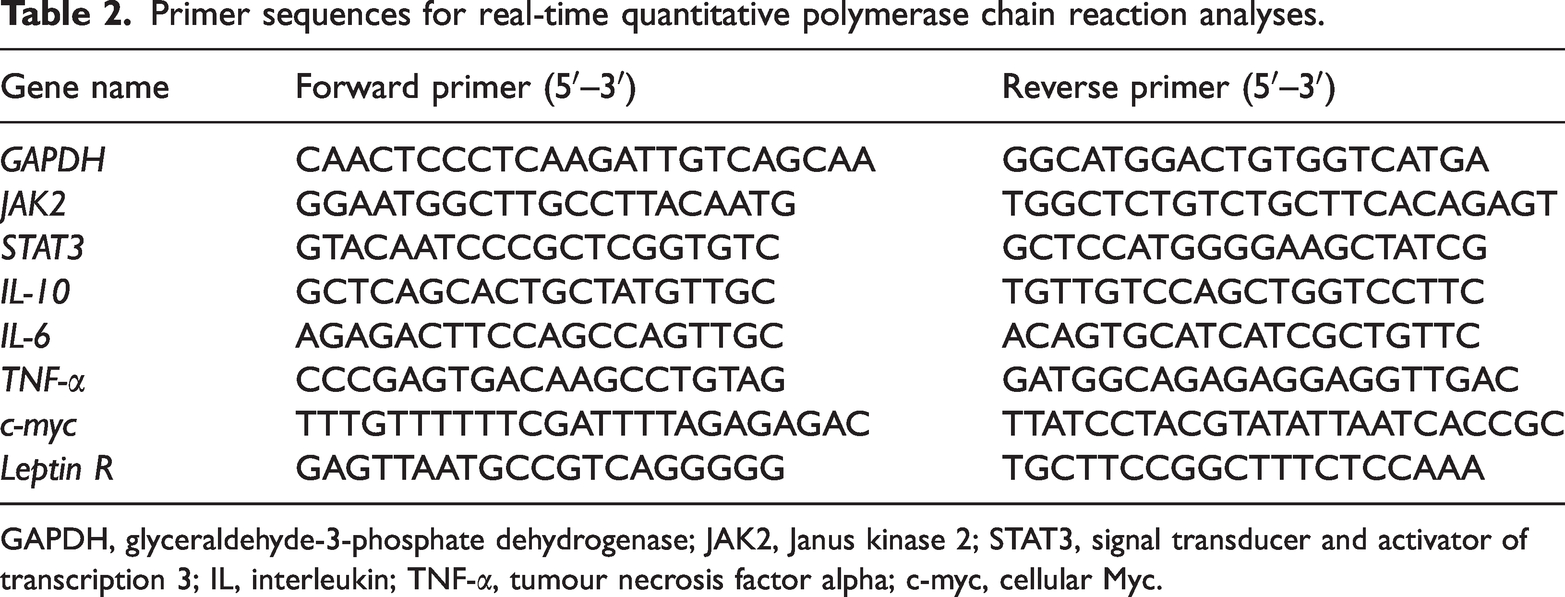
GAPDH, glyceraldehyde-3-phosphate dehydrogenase; JAK2, Janus kinase 2; STAT3, signal transducer and activator of transcription 3; IL, interleukin; TNF-α, tumour necrosis factor alpha; c-myc, cellular Myc.
Detecting LEPR promoter methylation level by methylated DNA immunoprecipitation (MeDIP)
An MeDIP analysis was undertaken according to the manufacturer's instructions (55009; Active Motif, Carlsbad, CA, USA). The entire genomic DNA was sonicated to nucleic acid fragments with lengths ranging from 200 base pair (bp) to 1000 bp. Single-stranded DNA was generated by the heat denaturation of 1 μg of fragmented DNA. Immunoprecipitation was performed at 4°C overnight using 1 μg anti-5-methylcytosine (5mC) antibody (Active Motif). Finally, the DNA fragments bound to the anti-5mC antibody were captured, eluted and used for real-time PCR detection using primers that target the LEPR gene promoter region.
Statistical analyses
All statistical analyses were performed using GraphPad Prism 9.01 (Graphpad Software Inc., San Diego, CA, USA). The experimental data were compiled and entered into Excel 2019 software (Microsoft Corporation, Redmond, WA, USA). Data are presented as mean ± SE. The comparison between two groups was undertaken using a two-tailed unpaired t-test and the comparison between three groups was undertaken using one-way analysis of variance. A P-value <0.05 was considered statistically significant.
Results
Leptin exerts a dose-dependent inhibitory effect on ischaemia–reperfusion injury
Different leptin concentrations (0 ng/ml, 50 ng/ml, 100 ng/ml, 200 ng/ml and 300 ng/ml) were used to pretreat PC12 neuronal cells to establish the OGD cell model. The viability of the PC12 neuronal cells was determined using a CCK-8 cytotoxicity assay after different reperfusion times (1 h, 6 h, 12 h and 24 h). PC12 neuronal cell viability was restored by 200 ng/ml leptin pretreatment (Figure 1(a)). PC12 cell viability was highest after 1 h of oxygen-glucose deprivation followed by reperfusion for 24 h in cells pretreated with 200 ng/ml leptin (Figure 1(b)).

(a) PC12 neuronal cell viability under different leptin concentrations (50–300 ng/ml) for 30 min after the OGD for 1 h and then the reperfusion of 24 h; (b) PC12 neuronal cell viability following different reperfusion times (1 h, 6 h, 12 h and 24 h) subsequent to pretreatment with different leptin concentrations (50–300 ng/ml) and (c, d) mRNA levels of the inflammatory factor interleukin (IL)-6 and the anti-inflammatory factor IL-10 relative to levels of GAPDH. Data were analysed using one-way analysis of variance; *P < 0.05; ***P < 0.001; compared with the control group. GAPDH, glyceraldehyde 3-phosphate dehydrogenase. The colour version of this figure is available at: http://imr.sagepub.com.
The optimal reperfusion time in PC12 neuronal cells was determined so that levels of mRNA of the inflammatory interleukin factor (IL)-6 and the anti-inflammatory factor IL-10 after pretreatment with different concentrations of leptin (0 ng/ml, 50 ng/ml, 100 ng/ml and 200 ng/ml) prior to 1 h of oxygen-glucose deprivation and 24 h of reperfusion could be determined using real-time quantitative PCR. The results demonstrated that an increase in the concentration of leptin significantly downregulated the levels of IL-6 mRNA (Figure 1(c); P = 0.015 compared with the control group) and significantly upregulated the levels of IL-10 mRNA (Figure 1(d); P = 0.0195 compared with the control group).
Leptin alleviates the extent of brain tissue injury
The area of cerebral infarction in rats after 1 h of ischaemia followed by reperfusion for 24 h was measured using TTC staining (Figures 2(a) & 2(b)). The area of cerebral infarction was significantly reduced in the group of rats that received leptin (MCAO + LP) pretreatment intraperitoneally compared with the saline-treated MCAO control group (P < 0.01). The areas of cerebral ischaemic injury were principally distributed in the blood supply region of the middle cerebral artery, including the prefrontal cortex and striatum (Figures 2(c) & 2(d)). Leptin administration reduced the ischaemic injury area of brain tissue in the region surrounding the cerebral infarction. There was better structural integrity in the leptin-treated group (MCAO + LP) at an identical location of the brain tissue compared with the saline-treated MCAO control group (Figure 2(e)).
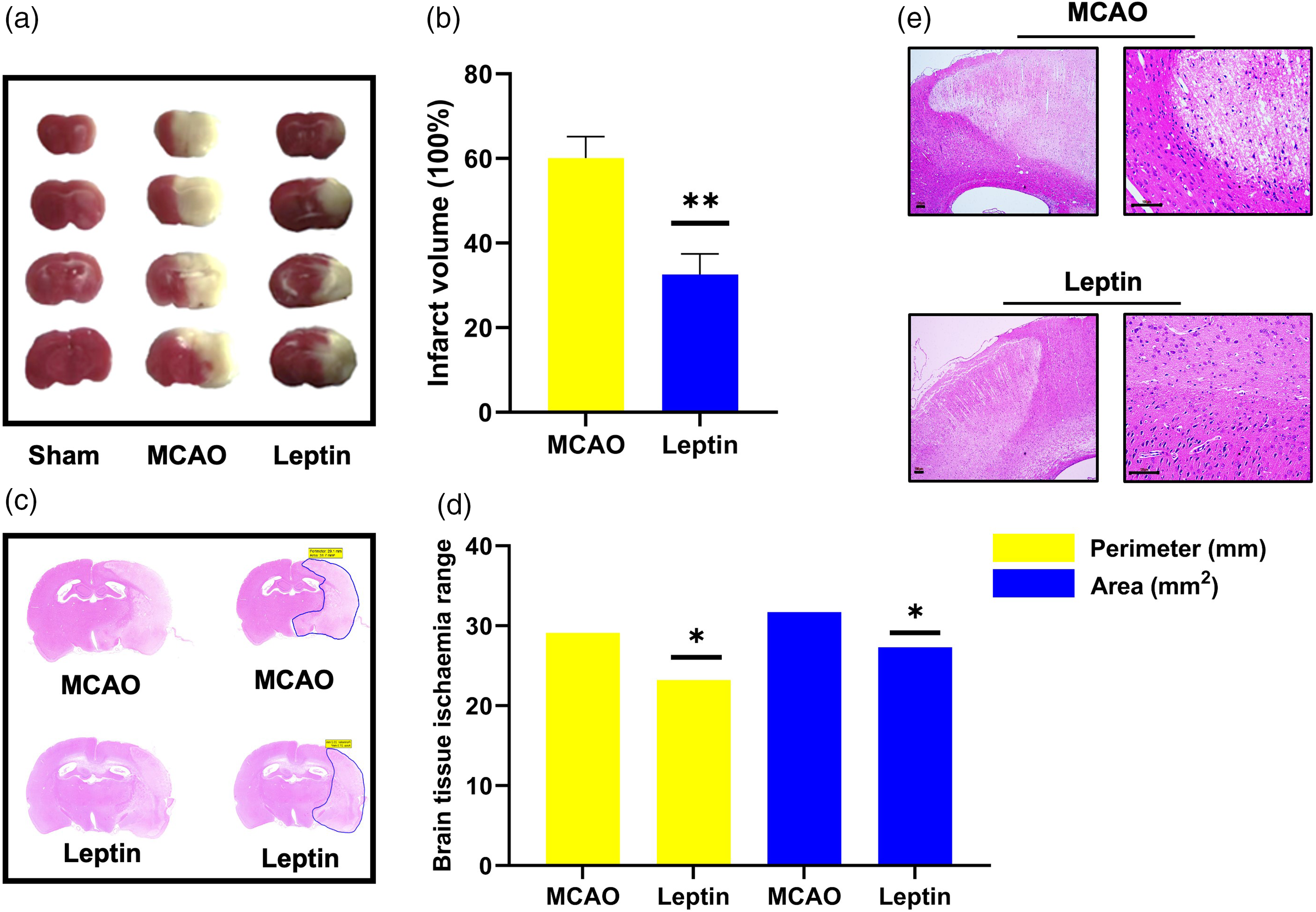
Ischaemic area analysis of the rat brain tissue by 2% 2,3,5-triphenyltetrazolium chloride staining after 1 h of ischaemia followed by 24 h of reperfusion (a, b). Ischaemic area analysis of the rat brain tissue in the coronal sections using haematoxylin and eosin staining with the brain tissue ischaemia range referring to the perimeter and area of ischaemic regions of brain tissue defined by blue lines on Figure 2(c) (c, d). Integrity of the brain tissue at the infarct region as observed under the microscope using haematoxylin and eosin staining (scale bar 100 µm) (e). The data were measured three times and analysed statistically using a two-tailed unpaired t-test or analysis of variance; *P < 0.05; **P < 0.01, compared with the MCAO control. SHAM, sham-operated group; MCAO, middle cerebral artery occlusion group; Leptin, MCAO group pretreated with leptin. The colour version of this figure is available at: http://imr.sagepub.com.
JAK2/STAT3 phosphorylation indicates leptin-mediated cerebroprotective effects
After establishing the MCAO rat model, real-time quantitative PCR analysis was used to measure the levels of IL-6, IL-10, tumour necrosis factor (TNF)-α, JAK2 and STAT3 mRNA in brain tissues from each experimental group of rats. Leptin pretreatment significantly upregulated the levels of the anti-inflammatory factor IL-10 (Figure 3(a), P < 0.001) compared with the MCAO group. The levels of the inflammatory factors IL-6 (Figure 3(b), P < 0.001) and TNF-α (Figure 3(c), P = 0.008) were significantly downregulated by leptin treatment compared with the MCAO group.
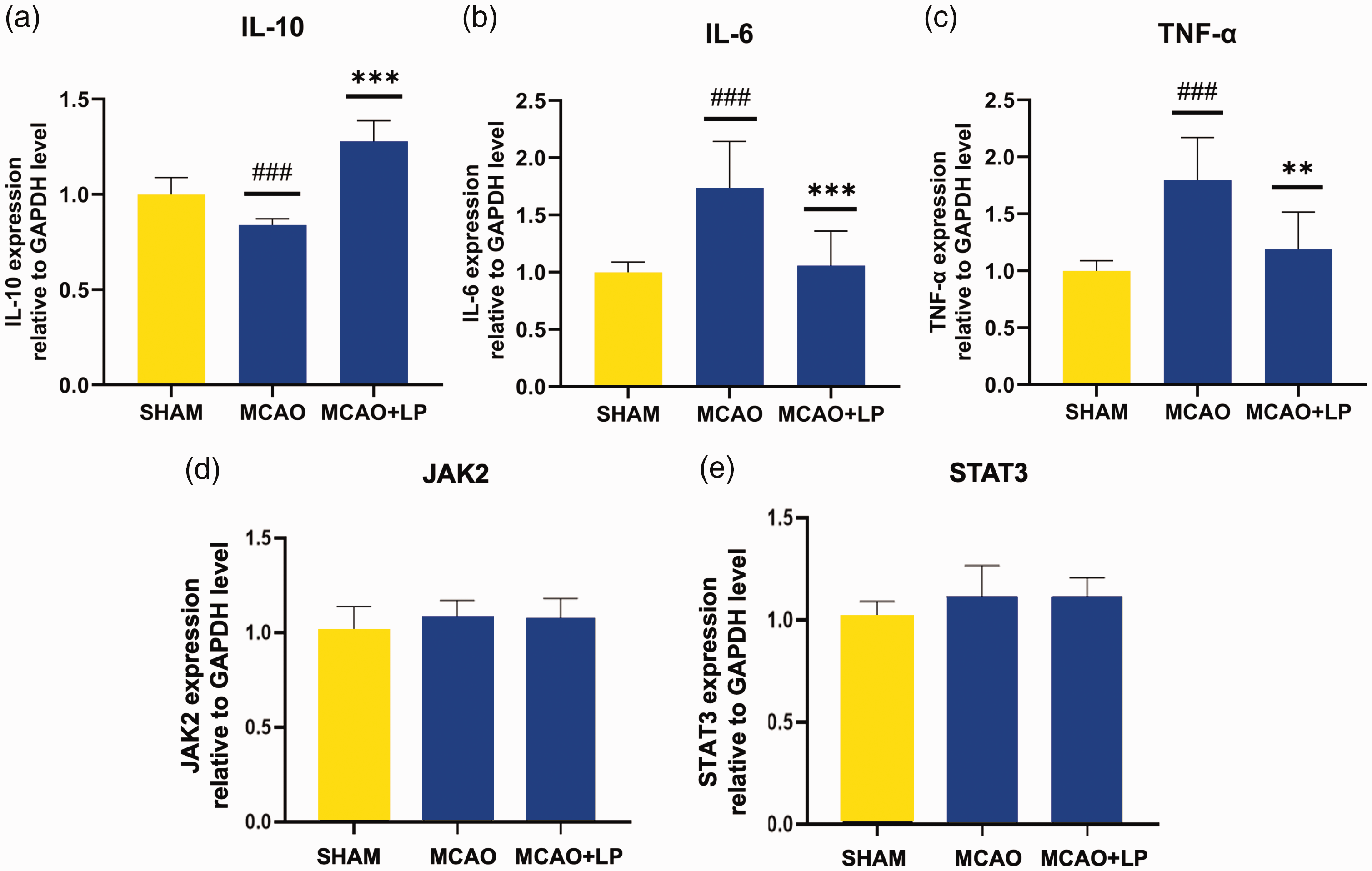
The levels of interleukin (IL)-10, IL-6, tumour necrosis factor (TNF)-α, JAK2 and STAT3 mRNA relative to levels of GAPDH in the brain tissues (a–e). The data were analysed using one-way analysis of variance; ###P < 0.001 compared with the SHAM group; **P < 0.01, ***P < 0.001, compared with the MCAO group. JAK2, Janus kinase 2; STAT3, signal transducer and activator of transcription 3; reverse transcription-quantitative polymerase chain reaction; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; SHAM, sham-operated group; MCAO, middle cerebral artery occlusion group; MCAO + LP, MCAO group pretreated with leptin. The colour version of this figure is available at: http://imr.sagepub.com.
The JAK2 and STAT3 proteins can have phosphorylated forms so these were measured using Western blot analysis. The results demonstrated a significant increase in the protein levels of p-STAT3 (Figure 4(a), P = 0.007), p-JAK2 (Figure 4(b), P = 0.008) and the anti-apoptotic protein B-cell leukaemia/lymphoma-2 (BCL-2) (Figure 4(c), P = 0.009) in response to leptin pretreatment (MCAO + LP) compared with the MCAO group. Furthermore, there was a significant decrease in the level of apoptotic protein caspase 3 protein in the leptin-pretreated group compared with the MCAO control group (Figure 4(d), P = 0.037).
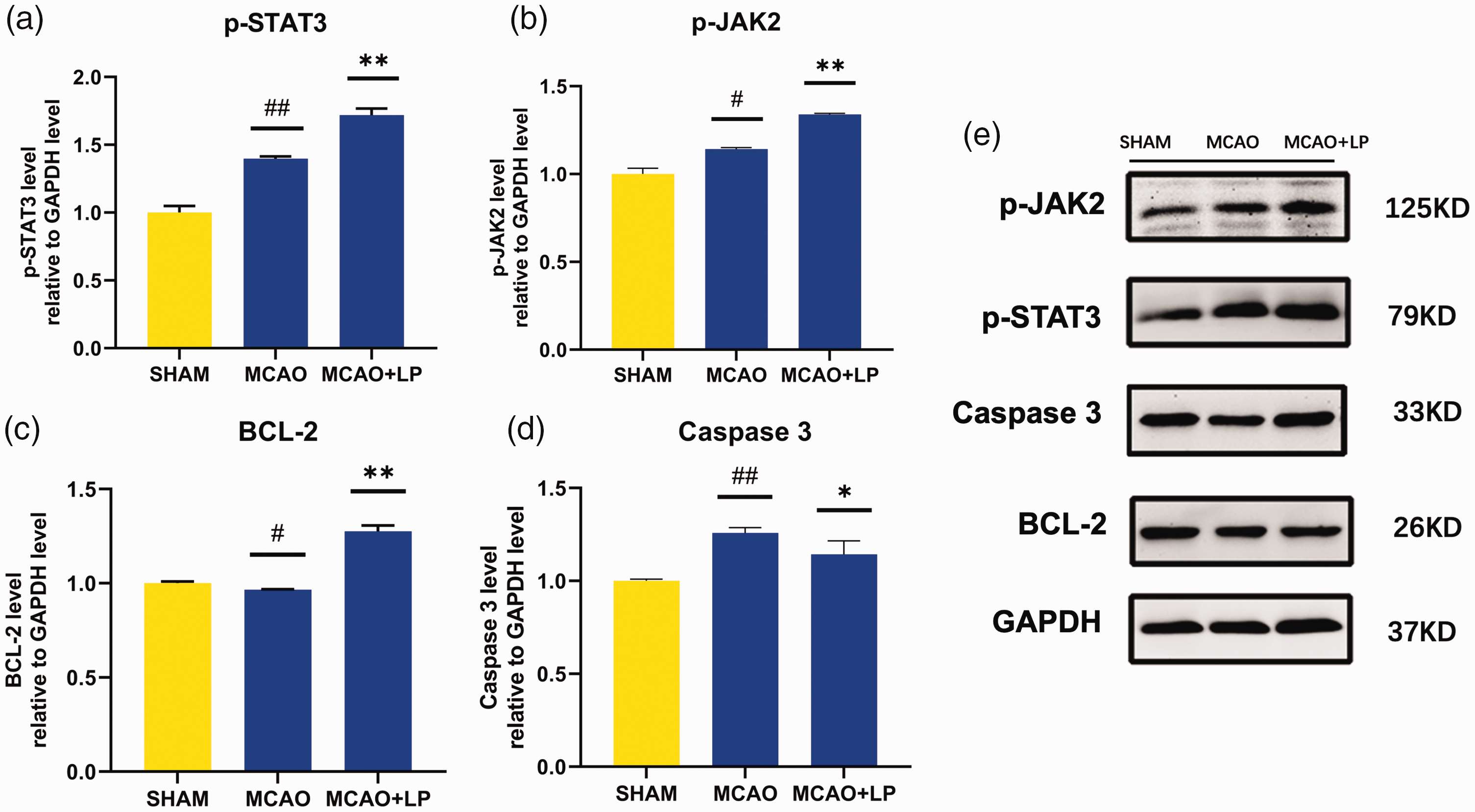
Western blot analysis showing the protein levels of phosphorylated-STAT3 (p-STAT3) (a), phosphorylated-JAK2 (p-JAK2) (b), anti-apoptotic protein BCL-2 (c) and apoptotic protein caspase 3 (d) relative to levels of GAPDH. Western blot analysis showing the levels of P-JAK2, P-STAT3, caspase 3, BCL-2 and GAPDH in the SHAM, MCAO and MCAO + LP groups. GAPDH was used as a loading control (e). The data were analysed using one-way analysis of variance; #P < 0.05, ##P < 0.01, compared with the SHAM group; *P < 0.05, **P < 0.01, compared with the MCAO group; STAT3, signal transducer and activator of transcription 3; JAK2, Janus kinase 2; BCL-2, B-cell lymphoma 2; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; SHAM, sham-operated group; MCAO, middle cerebral artery occlusion group; MCAO + LP, MCAO group pretreated with leptin. The colour version of this figure is available at: http://imr.sagepub.com.
Leptin pretreatment alters the methylation level of the LEPR promoter region
The level of LEPR mRNA was significantly higher within the ischaemic penumbra of the leptin pretreated group (MCAO + LP) compared with the saline-pretreated MCAO control group (Figure 5(a), P = 0.009). The leptin pretreated group (MCAO + LP) exhibited significantly decreased total 5mC levels in the brain tissue of the ischaemic penumbra region compared with the saline-pretreated MCAO control group (Figure 5(b), P = 0.042). The leptin pretreated group (MCAO + LP) demonstrated a significantly lower methylation level of the LEPR gene promoter region compared with saline-pretreated MCAO control group (Figure 5(c), P = 0.047).
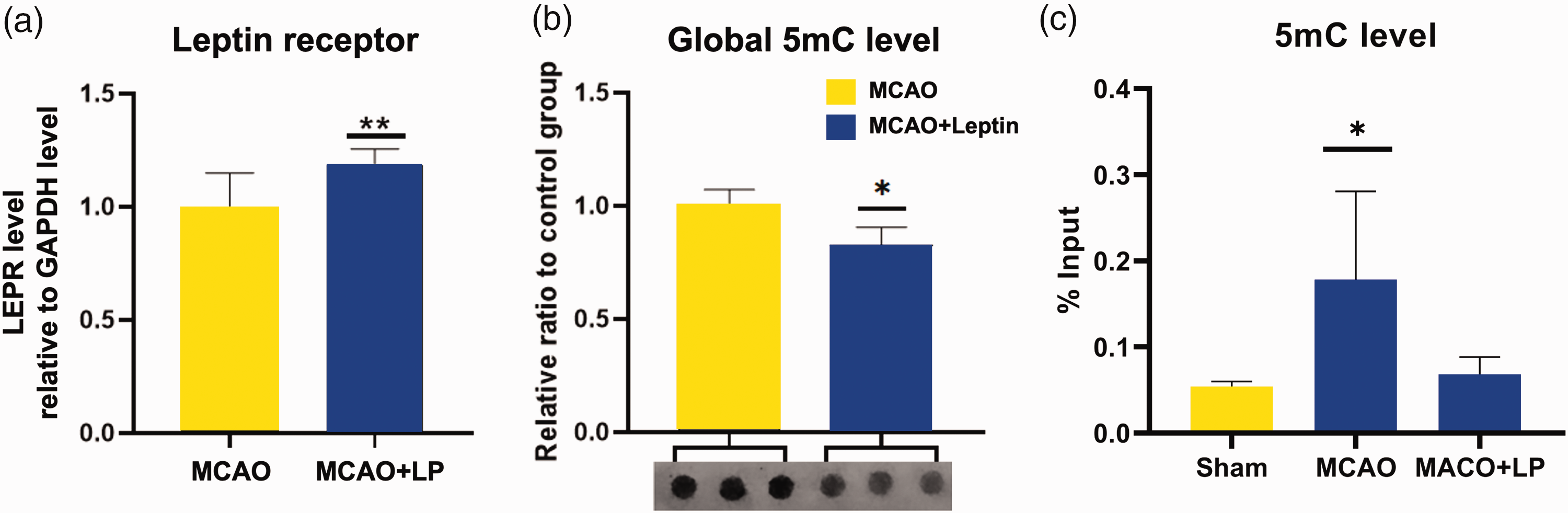
Quantitative results of the mRNA levels of leptin receptor (LEPR) relative to levels of GAPDH (a). Dot blot experiment showing the DNA methylation level of the genomic DNA (global 5-methylcytosine [5mC]) (b). DNA methylation level of the LEPR gene promoter region 5mC (c). The data were analysed statistically using a two-tailed unpaired t-test or analysis of variance; *P < 0.05; **P < 0.01; comparisons between the MCAO and MCAO + LP groups. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; Sham, sham-operated group; MCAO, middle cerebral artery occlusion group; MCAO + LP, MCAO group pretreated with leptin. The colour version of this figure is available at: http://imr.sagepub.com.
Discussion
While the neuroprotective effect of leptin through the JAK2/STAT3 pathway has been previously reported in both in vitro and in vivo models of cerebral ischaemia,19,22 this current study offers an enhanced understanding of this effect through four aspects: (i) the optimal leptin concentration for exerting neuroprotective effects against ischaemia–reperfusion injury was 200 ng/ml in vitro, but excessive leptin diminished this effect; (ii) leptin pretreatment in the MCAO rat model demonstrated a similar effect to leptin administration post-CIRI. This finding suggests a novel approach to utilizing leptin during cerebral ischaemia injuries; (iii) in addition to regulating the expression of inflammation-related factors, leptin also impacts the levels of proteins related to apoptosis. The current Western blot analysis showed that leptin pretreatment led to upregulated levels of the anti-apoptotic protein BCL-2 and downregulated the levels of the apoptotic protein caspase 3; (iv) the MeDIP analysis demonstrated that DNA methylation regulated LEPR gene expression in the MCAO rat model of cerebral ischaemia injury when leptin pretreatment was used. This finding expands our understanding of the intricate interactions that underpin this biological process.
Impaired energy metabolism results in cerebral ischaemia–reperfusion injury.23–25 The primary function of leptin is to regulate energy metabolism. Simultaneously, it exerts numerous biological effects, such as improving insulin resistance, promoting fatty acid oxidation and having anti-inflammatory, anti-atherosclerotic and anti-apoptotic effects.26–28 Furthermore, leptin is closely related to the incidence of cardiovascular disease. 7 Researchers have reported a correlation between leptin and cardiovascular diseases, with leptin being cardioprotective in animals both in vivo and in vitro. 7 However, the research and clinical application of leptin in cerebrovascular diseases is at an early stage. Moreover, leptin exerts contradictory and diverse effects, which may be related to changes in the expression of LEPR-related signals. Previously,4–6 leptin was reported to be associated with plaque instability in patients with high levels of leptin antigen-antibody response or LEPR-induced inflammation, which commonly leads to stroke and transient ischaemic attack. However, leptin effectively reduces neuronal intracellular BCL-2 and caspase 3 expression in injury-induced neuronal cells, thereby exerting neuroprotective effects through its anti-apoptotic function.23,29,30 A previous study injected 0.1 mg/kg leptin in the lateral ventricle of dogs 30 min before ischaemia–reperfusion surgery. 8 The protective effect of leptin on neurons in the cerebral ischaemic region could reach a maximum during this time and this effect was independent of the changes in cerebral blood flow.10,31,32 The current study explored a new leptin pretreatment method, which was a daily intraperitoneal injection for 1 week in advance of the ischaemia–reperfusion injury. This method not only reduces the operational difficulty of drug delivery via a lateral ventricle injection but also exerts similar cerebral protective effects after leptin treatment. The MCAO animal model constructed in rats confirmed that the neuroprotective effect of leptin in CIRI injury was consistent with its role in rats, thus establishing the universal brain protective effect of leptin. In a neuronal OGD cell model established using the PC12 cell line, the current study determined that the optimal drug concentration and reperfusion time for leptin pretreatment. 33 For a leptin concentration of 200 ng/ml and reperfusion time of 24 h, the PC12 neuronal cells demonstrated the highest cellular viability. This finding was consistent with the results of the OGD cell model constructed using rat metaplastic neurons in other research. 29 Therefore, drug toxicity offsets the neuroprotective effect of leptin for a concentration > 200 ng/ml, which provides reference information for exploring the appropriate leptin concentration. These findings suggest that, in certain instances, leptin has the potential to worsen tissue injury.
Leptin has demonstrated efficacy in alleviating ischaemia–reperfusion injury by suppressing inflammatory responses and averting apoptosis across various organs, including the liver, intestine and ovary. 7 Conversely, studies in obese rats have revealed a contrasting phenomenon, where localized leptin aggregation around cardiomyocytes induces apoptosis. 19 This localized effect occurs adjacent to pericardial adipose tissue and operates through both the JAK2/ROS/NKA and JAK2/ROS/mitochondria pathways. 19 Additionally, elevated leptin levels can lead to receptor desensitization, diminishing responsiveness to its physiological functions. 19 Mechanisms such as receptor downregulation and impaired signal transduction pathways contribute to this desensitization, potentially leading to metabolic dysregulation or leptin resistance, commonly observed in obesity-related studies. 19 The observed adverse effects associated with high leptin doses underscore the necessity for meticulous dosage titration to balance therapeutic benefits against potential side-effects. Further research is warranted to elucidate the precise dose-dependent mechanisms of leptin and to optimize its therapeutic utility in neurological disorders such as stroke.
While constructing the rat MCAO model in the current study, the toxic effect of high leptin concentrations was taken into consideration. A dose of 200 ng/ml leptin was injected intraperitoneally daily for 1 week in advance of the ischaemia–reperfusion injury, which totalled 20 µl. The body weight of the rats was controlled between 250 g and 280 g, which facilitated maintaining leptin blood concentrations from approximately 180 ng/ml to 200 ng/ml. This process ensured that the study could assess the predominant neuroprotective effects of leptin in rats. The loss of leptin caused by the action of the blood–brain barrier was not calculated. Furthermore, the actual leptin concentration that entered across the blood–brain barrier of rats was unknown. Because of the limited research time, the current study did not address this specific issue. However, at a leptin pretreatment concentration of 200 ng/ml and a reperfusion time of 24 h, the leptin-treated group demonstrated a significant reduction in the area of ischaemic brain tissue compared with the control group. Furthermore, H&E staining results of the brain tissue sections demonstrated a higher degree of integrity of the ischaemic brain tissue and a lower degree of damage. Therefore, the cerebroprotective effect of leptin remained and was higher at a concentration of 200 ng/ml. The overall levels of JAK2 and STAT3 within the leptin-activated JAK2/STAT3 pathway remained unchanged. Notably, the changes were confined to an elevation solely in the phosphorylated forms of JAK2 and STAT3. This current finding warrants further investigation into the factor that induces the increased phosphorylation levels of JAK2 and STAT3 in the leptin pretreatment group. This current study focused on the upstream component of JAK2, namely the LEPR. PCR results unequivocally substantiated an augmented LEPR levels in the leptin pretreatment group.
Epigenetics is the study of the complex interaction between genes and their products that determine the phenotype. DNA methylation has emerged as a focal point in epigenetic research because of its early recognition and in-depth exploration.34–36 This epigenetic modification intricately regulates gene expression, chromosome inactivation, repetitive sequence repression and carcinogenesis. Reduced DNA methylation levels are correlated with elevated gene expression. 37 In the current study, DNA immunoprecipitation experiments specifically targeting the methylated fragments within genomic DNA from ischaemic regions demonstrated a significant decrease in the genomic DNA methylation levels within ischaemia-affected brain tissues. After leptin pretreatment, the increased levels of LEPR mRNA presents an intriguing phenomenon, the underlying reasons for which remain elusive. These current findings suggest that this phenomenon could be linked to alterations in methylation levels. The current study also measured changes in the methylation level of the LEPR gene promoter region to determine whether the LEPR gene was demethylated while genomic methylation levels were reduced. The MeDIP analysis demonstrated reduced methylation levels in the LEPR gene promoter region after leptin pretreatment, which would promote the expression of the LEPR gene, thereby increasing the number of LEPRs on the cell membranes. Upon binding to these receptors, leptin activates downstream signalling molecules, namely JAK2 and STAT3, through phosphorylation, exerting a cerebral transthoracic protective effect. However, future research should determine the duration of this methylation level change after the cessation of leptin treatment.
In conclusion, exogenous leptin might bind to extra-activated LEPR by reducing the methylation level of the LEPR gene promoter region, which leads to an increase in phosphorylated JAK2/STAT3 and apoptotic signalling pathways. Therefore, exogenous leptin can play a role in anti-inflammation and anti-apoptosis, thus protecting damaged brain tissue from reperfusion.
Footnotes
Acknowledgement
We thank Bullet Edits Limited for the linguistic editing and proofreading of the manuscript.
Author contributions
Wenyuan Zhao: conceptualization, methodology; Sha Liu: conceptualization, formal analysis; Xuelou Wang: investigation, formal analysis, writing; Zhen Wang: investigation, formal analysis, writing; Yu Feng: resources, investigation; Tingbao Zhang: formal analysis, writing; Zhongxiang Wu: writing; Junjie Huang: methodology.
Data availability statement
The raw data are publicly available upon request. The corresponding author Wenyuan Zhao can be contacted at
Declaration of conflicting interest
The authors declare that there are no conflicts of interest.
Funding
This research was funded by the Technology Innovation Special Major Project of Hubei Province (Grant Number 2022BAC003).