
Abstract
Objectives
Mycosis fungoides (MF) is the most common cutaneous T-cell lymphoma; it arises from tissue-resident memory T-cells (TRM). In the present study, we investigated potential functional genetic variations that may predispose MF development.
Methods
A case–control study was conducted using whole-exome sequencing, with a focus on genes that are essential to TRM function.
Results
We included 21 patients and 19 healthy subjects in the study. Single nucleotide polymorphisms in the following genes were significantly more common in patients than in healthy subjects: GZMB, HLA-DRB1, CD103, and NOTCH1. Moreover, the number of patients carrying single nucleotide polymorphisms in LAG3, NR4A2, and CD26L was significantly greater in the patient group than in the control group.
Conclusions
The presence of genetic variations in one or more TRM functional gene may predispose patients to develop MF. Further studies involving a larger patient population and a comparative analysis of protein expression will be necessary to validate these findings.
Keywords
Introduction
Mycosis fungoides (MF) is a mature T-cell lymphoma that arises in the skin. In MF, neoplastic lymphocytes exhibit hyperconvoluted nuclei and have a strong proclivity to infiltrate the epidermis. Clinically, the disease is characterized by a protracted stepwise evolution, from patches to plaques that may eventually progress to tumors. Disseminated disease occurs in a minority of patients. The peak age of incidence is in the late fifth decade, but MF can also arise in children.1,2
MF is the most common primary cutaneous lymphoma, representing 50% to 90% of all cases.1,3 The cells of origin of MF are thought to be CD45RO (receptor-type tyrosine-protein phosphatase C short isoform)-positive effector resident memory T-cells (TRM) that generally reside in the skin. These cells normally express molecules that facilitate their migration from the blood to the skin, such as C-C chemokine receptor (CCR)4, CCR6, and CCR7. Although the existence of several subtypes of effector memory T-cells—such as T-helper (TH)-1, TH-2, and TH-17 cells—may explain the heterogeneity of MF, they are not currently of any clinical significance. 4 Moreover, despite the investigation of the diagnostic significance of some TRM-related antigens in surgical pathology practice, they are not widely used in MF diagnosis. 5
The etiology of MF has yet to be determined, although several environmental and genetic factors have been related to the development of the disease. The objective of the present study was therefore to investigate genetic polymorphisms in a panel of genes that are related to TRM cell function.
Patients and methods
Study group
This retrospective study was conducted in the Departments of Pathology and Dermatology at Jordan University Hospital, a tertiary center in Amman, Jordan, between 2022 and 2023. The Scientific Research Committee at the School of Medicine, The University of Jordan (425/2022/67 on 23 January 2022), and the Institutional Review Board at Jordan University Hospital (10/2022/4248 on 17 February 2022) approved the study. Patients diagnosed with MF were consecutively recruited into the study. The control group comprised healthy subjects. Signed consent was obtained from all participants, and all participant information was de-identified. The study size was arrived at by enrolling all known MF patients from our archives who were willing to participate, and then enrolling a similar number of control subjects.
The collected data included clinicopathological information regarding age, sex, clinical stage, disease duration, and therapy type. The reporting of this study conforms to Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) guidelines. 6
Whole-exome sequencing
Laboratory specialists collected whole blood (20 mL) from all participants into ethylenediaminetetraacetic acid-containing tubes. DNA extraction was performed using the Zymo Quick-DNA kit (Zymo Research Corporation, Irvine, CA, USA) following the manufacturer’s instructions. The extracted DNA samples were outsourced to Macrogen Inc. (Seoul, South Korea). Sequencing reads were aligned to GRCh37 using the BWA-mem algorithm. Variant classification and annotations were performed using the VarSeq analysis workflow (Golden Helix, Bozeman, MT, USA), implementing the guidelines of the American College of Medical Genetics and Genomics.
Variant assessment process
The impact of each identified variant in the context of human disease was annotated and evaluated using the following databases and in silico algorithms: 1000 Genomes, Genome Aggregation Database (gnomAD), ClinVar, Online Mendelian Inheritance in Man (OMIM), National Center for Biotechnology Information SNP Database (NCBI dbSNP), NCBI Reference Sequence database (RefSeq Genes), Exome Aggregation Consortium (ExAC) Gene Constraints, Sorting Intolerant From Tolerant (VS-SIFT), Polymorphism Phenotyping v2 (VS-PolyPhen-2), PhyloP, GERB++, GeneSplicer, MaxEntScan, Splice Site Prediction by Neural Network (NNSplice), and Position Weight Matrix (PWM) Splice Predictor. The analysis was reported using Human Genome Variation Society nomenclature, as implemented by the VarSeq transcript annotation algorithm. The reported transcript style matches that used most frequently by the clinical labs who submit to ClinVar.
Variants were excluded if they had a low putative impact, had downstream or upstream genes, caused synonymous or silent effects, or were located in intergenic or 3′- or 5′-untranslated regions.
The SIFT algorithm was used to predict the impact of amino acid alterations on protein function and distinguish between deleterious variants (causing functional changes) and tolerated variants (with minimal impact). 7 The resulting scores, ranging from 0.0 to 1.0, were used to classify variants as deleterious (scores below 0.05) or tolerated.
Identifying genes of interest
The present study focused on 41 genes that are critical for TRM cell function. 8 Single nucleotide polymorphisms (SNPs) were assessed in the following genes: RUNX3, NOTCH1, PRDM1, ZNF683, NR4A2, JUNB, FOSL2, S1PR1, CD26L, CCR7, ITGA1, CD69, ITGA4, CD44, EGR2, KLF2, HIF1A, TGFB1, CD103, ITGB7, PDCD1, HACR2, CTLA4, LAG3, IFNG, CXCR6, NR4A1, IL7R, CXCR6, NR4A1, IL7R, CXCR3, SELP, CD244, TNFRSF9, GZMA, HLA-DRB1, RGS1, TOX1, GZMB, and CD45RO. The corresponding gene IDs are available in Supplementary table 1.
Statistical analysis
The SciPy package (version 1.11.1) in Python was used to analyze the results. 9 Continuous and nominal variables are described as means and counts (frequencies), respectively. Descriptive statistics were calculated for assessment at 95% confidence intervals based on binominal distribution. Statistical differences between patient and control groups were analyzed using a two-sided Fisher’s exact test for variant assessments. A significance level of P < 0.05 was applied.
Results
Clinical characteristics
In the current study, 21 patients were enrolled, comprising 10 male patients (47%) and 11 female patients (52%) with ages ranging from 5 to 85 years (mean 43 years). All patients presented with stage I disease; five patients (24%) had stage IA and 16 patients (76%) had stage IB. The disease duration (from time of diagnosis) varied from 0.12 to 14 years, with a mean duration of 4 years. All patients were treated with topical steroids, and 18 patients (86%) underwent additional phototherapy. All patients had a stable clinical course and none showed disease progression or fatal outcome. The control group consisted of 19 healthy subjects, comprising 11 male subjects (58%) and eight female subjects (42%) with ages ranging from 8 to 71 years (mean 32 years).
SNPs in genes of interest
The patient dataset contained 2871 SNP variants, and there were 1942 variants in the control group. The numbers of SNPs in GZMB (P = 0.0002), HLA-DRB1 (P < 0.0001), CD103 (P = 0.0213), and NOTCH1 (P = 0.002) were significantly higher in patients than in control subjects (Figure 1). Moreover, significantly more patients than control subjects carried SNPs in LAG3 (P = 0.0088), NR4A2 (P = 0.042), and CD26L (P = 0.018). Although SNPs in CXCR6 and S1PR1 were detected in patients only, they were not significantly different between the two groups (Figure 2).

Distribution of single nucleotide polymorphisms among patients (blue bars) and control subjects (gray bars). P-values are represented within the bars.

Distribution of genes of interest carrying single nucleotide polymorphisms among patients (blue bars) and control subjects (gray bars). P-values are represented within the bars.
The number of SNPs in ITGB7 was significantly higher in the control group than in the patient group. Moreover, one healthy individual carried a single SNP variant in IFNG that was not detected in any patient. JUNB, CXCR3, EGR2, and HACR2 did not meet the inclusion criteria determined by the American College of Medical Genetics and Genomics guidelines. Table 1 includes details of the results.
Distribution of single nucleotide polymorphisms (SNPs) in genes related to tissue resident T-cell function among patients with mycosis fungoides and control subjects.

Significant values are shown in red.
We further investigated the variants in the following genes that had significant P-values: NOTCH1, GZMB, CD103, HLA-DRB1, CD26L, NR4A2, LAG3, ITGB7, S1PR1, and CXCR6. The total number of variants in these genes was 1132, the majority of which were predicted as intronic (924 variants); 208 variants were predicted as exonic. The majority of the exonic variants were missense (204), and only four were frameshift mutations (Figure 3).

Variant effects among selected genes.
Prediction of variant pathogenicity
The SIFT algorithm did not predict any amino acid changes in 928 of the detected variants. However, it predicted 44 deleterious variants and 160 tolerated variants. The deleterious variants were spread among CD103 and HLA-DRB1 (38 and 6 variants, respectively) (Table 2). The variants predicted to be deleterious or tolerated were identified as missense variants, whereas in those with unknown effects, 924 were intronic and four were identified to cause a frameshift. Furthermore, all identified frameshift variants existed within HLA-DRB1. Notably, all NR4A2 variants were intronic, and LAG3 contained only missense variants.
Distribution of Sorting Intolerant From Tolerant (SIFT) predictions by gene.

Investigating deleterious variants
The 44 deleterious variants included 6 known SNPs (dbSNP138 IDs: rs71366574, rs1716, rs17887154, rs3744679, rs2272606, and rs1064592) that were detected in HLA-DRB1 and CD103 (transcript IDs: NM_002208.5 and NM_002124.3, respectively). These deleterious variants were identified in 21 patients and were all frameshift mutations with a moderate putative impact.
According to the NCBI dbSNP, the clinical significance of almost all of these variants is unknown, except for the rs2272606 SNP; this was interpreted as benign with a one-star review in a single publication. 10 In our series, deleterious variants did not have any significant clinical associations with sex, age (>50 vs. <50 years) or stage (IA vs. IB).
Allele frequencies in different populations
Allele frequencies in different populations were investigated to assess variance correlations in specific ethnic groups (Figure 4). In the dbSNP, there were unfortunately no data regarding allele frequencies in West Asian or Middle Eastern populations. The NOTCH1 median allele frequency was above 0.5 in all tested populations, whereas the CD103 median allele frequency was above 0.5 in African populations only.
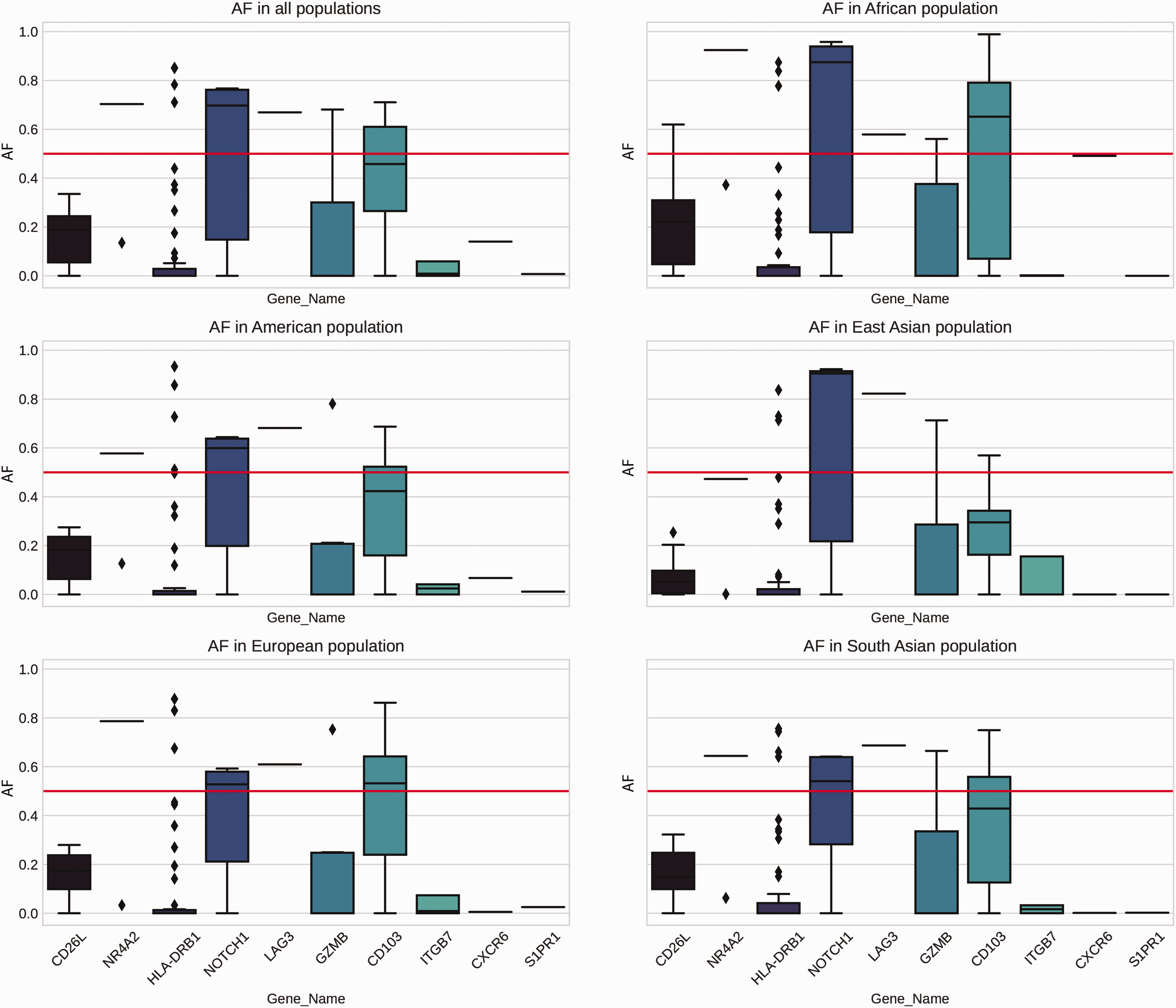
Allele frequencies (AFs) of selected genes among different demographic populations.
Discussion
MF is the most common cutaneous lymphoma, comprising 55% to 90% of all cases.1,3 Clinically, most patients have patches and plaques with an indolent clinical course, although one-quarter have disease progression and dissemination. The disease is more common in men than in women and has a peak incidence in the late fifties. Histological examinations, immunohistochemical studies, and clinicopathological correlations remain the optimal diagnostic modalities. 4 Genetic aberrations are common in MF, such as mutations in TP53; increased expression of NA3, JUNB, and c-MYC; a loss of tumor suppressors CDKN2A and CDKN2B; hypermethylation of mismatch repair genes; and dysregulation of Janus kinase/signal transducer and activator of transcription protein pathways. 11 However, these alterations are acquired and are not consistently present in MF.
Epidemiological studies have revealed a steady increase in MF incidence in the last three decades, especially in industrialized countries.12,13 Although no clear etiological factors have been identified, it is hypothesized that the persistent activation of cutaneous TRM by diverse environmental agents, along with failed cellular apoptosis, results in malignant transformation. This hypothesis is supported by the observation that MF is often preceded by non-specific dermatitis and has a prolonged indolent clinical course before transformation. 1
Chronic occupational exposure to solvents—particularly halogenated hydrocarbons, pesticides, plastics, metals, or cutting oils—appears to be associated with MF development.14–16 Furthermore, solar radiation, tobacco smoking, and alcohol are high-risk environmental and lifestyle factors for MF, especially when combined.17–19 This might explain the higher frequency of MF among men because they are more likely than women to be exposed to the aforementioned jobs and risk factors. Other reported risk factors include drugs and microbial infections; however, it is believed that these induce lymphocyte proliferation, thus uncovering a hidden malignant clone of cells, rather than being independent carcinogenic factors.1,5,20
Genetic predispositions have been implicated in MF development. For example, the familial clustering of cases was described in early reports.21,22 Similarly, certain alleles of human leukocyte antigens (HLA) have an influence on the susceptibility and prognosis of MF. It is hypothesized that genetic polymorphisms in HLA genes may influence the ability of the immune system to recognize and eliminate cancer cells.23–25 Similarly, our study revealed an association between MF and HLA-DRB1 polymorphisms. In addition, several studies have noted the role of polymorphisms in genes related to inflammation and lymphocyte activation, such as COX2, IL2, IL13, IL17, STAT3, miRNAs, CTLA4, TGFB1, and VDR.26–34 However, other studies have discounted any association between MF and CTLA4, TGFB1, or VDR.35–38 Similarly, our findings did not support any influence of CTLA4 on MF. The link between MF and the aforementioned genetic polymorphisms thus remains controversial.
The genes investigated in the present study have various functional roles in TRM cells under normal circumstances. For example, CD103, CD26L, and CXCR6 are biomarkers of cancer-related-TRM cells, and the mobility of these cells is promoted by S1PR1 and CD26L. Moreover, the cytotoxic role of TRM cells is mediated by ITGB7, GZMB, HLA-DRB1, NOTCH1, and NR4A2, whereas LAG3 and CD103 serve as immune checkpoints and have anti-tumor effects. 8
To the best of our knowledge, this is the first study to examine a large panel of genes that are directly crucial for the function of TRM cells. Frequent genetic variations were identified in several genes, and deleterious effects were not uncommon—especially in HLA-DRB1 and CD103, which were identified in the MF patient group only. Although the impact of CD103 variants on chronic inflammation is difficult to predict, CD103 deficiency in mouse models has been associated with spontaneous cutaneous inflammation as well as autoimmune diseases as the result of T-regulatory cell dysfunction.39–41 In addition, cluster of differentiation 103 (CD103) protein is reportedly essential for the ability of TRM to infiltrate and control the growth of several solid tumors, which is associated with favorable outcomes. The recent use of immunomodulatory therapies and checkpoint inhibitors has also had success in certain cancers by enhancing the effectiveness of CD103-expressing T-cells. Thus, abnormalities in CD103 may result in ineffective antitumor immune responses. 42
Similarly, there is accumulating evidence regarding the role of the HLA system in many tumors, and HLA-DRB1 has been reported as significantly associated with MF, which strongly suggests the potential role of HLA-DRB1 genetic polymorphisms in this disease. HLA-DRB1 is a subunit of the HLA class-II molecules that interact with CD4+ T-lymphocytes, including TRM cells. Variations in this gene may thus influence the regulation of immune responses to environmental factors and contribute to the balance between anti-tumor activity and tolerance.25,43,44 The scarcity of these variants among the general population and in different countries also support their influence on disease development.
The present study has some limitations. First, some of the detected variants have not yet been reported in the literature. Second, the cohort size was relatively small. Third, genetic testing of first-degree relatives was not performed (except for in one patient) to examine the presence of the identified SNPs among family members. Finally, protein testing was not carried out because of the large number of examined genes. Further confirmatory studies are therefore recommended.
In conclusion, the current study highlights the role of genetic predisposition in MF development, providing statistical evidence that SNPs in TRM-related genes occur more commonly among MF patients, which may influence the function of TRM cells. This altered function might result in exaggerated responses to environmental stimuli and potentially increase the transformation probability of these cells. Future studies that evaluate the functionality of relevant proteins are needed to provide additional evidence of these findings.
Supplemental Material
sj-zip-1-imr-10.1177_03000605241239034 - Supplemental material for Genetic predisposition to early mycosis fungoides: investigating genetic polymorphisms in tissue-resident memory T-cell genes
Supplemental material, sj-zip-1-imr-10.1177_03000605241239034 for Genetic predisposition to early mycosis fungoides: investigating genetic polymorphisms in tissue-resident memory T-cell genes by Nour Almaani, Fatima Farhan, Salsabiela Bani Hamad, Eman A Abuhawileh, Lana Koubaitary, Mamoun Ahram and Tariq N Aladily in Journal of International Medical Research
Supplemental Material
sj-pdf-2-imr-10.1177_03000605241239034 - Supplemental material for Genetic predisposition to early mycosis fungoides: investigating genetic polymorphisms in tissue-resident memory T-cell genes
Supplemental material, sj-pdf-2-imr-10.1177_03000605241239034 for Genetic predisposition to early mycosis fungoides: investigating genetic polymorphisms in tissue-resident memory T-cell genes by Nour Almaani, Fatima Farhan, Salsabiela Bani Hamad, Eman A Abuhawileh, Lana Koubaitary, Mamoun Ahram and Tariq N Aladily in Journal of International Medical Research
Footnotes
Author contributions
All authors actively contributed to the research. NA and SBH: source of patients, reviewing manuscript. EAA and LK: specimen collection and handling. FF: statistical analysis, producing results. MA: reviewing manuscript, data analysis. TNA: research idea, moderation, writing manuscript.
Declaration of conflicting interests
The authors declare that there is no conflict of interest.
Funding
The research was financially supported by a generous grant from the Scientific Research Support Fund of Jordan – Ministry of Higher Education and Scientific Research (project number MPH/1/57/2021).
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.