
Abstract
Objective
To investigate the roles and underlying mechanisms of vascular endothelial growth factor receptor-3 (VEGFR-3) in gastric cancer (GC).
Methods
VEGFR-3 gene expression profiles in human gastric adenocarcinoma (GAC) tissues were analysed using The Cancer Genome Atlas database. Human GC cell lines and were used for in vitro studies. Mouse models of GC and distant metastasis were used for in vivo studies. Silencing of VEGFR-3 gene expression was achieved using small interfering RNA.
Results
VEGFR-3 gene expression was significantly elevated in GAC tissues and GC cells. Higher VEGFR-3 expression was positively correlated with more advanced stages and a greater number of metastatic lymph nodes. In vitro studies in GC cells showed that knockdown of VEGFR-3 gene expression significantly suppressed cell proliferation and migration, but promoted apoptosis. In vivo investigations revealed that silencing of VEGFR-3 gene expression exhibited significant inhibition on tumour growth and metastasis. Further mechanistic studies showed that VEGFR-3 exerted its pathological roles by affecting the key molecules in the apoptotic and epithelial–mesenchymal transition pathways.
Conclusion
The molecular pathways associated with VEGFR-3-mediated pathological effects could be targets in the development of novel approaches for the diagnosis, prognosis and treatment of GC.
Introduction
Gastric cancer (GC) is a leading cause of cancer-related death throughout the world. 1 GC is categorized into histologically different types: adenocarcinoma, adenosquamous cell carcinoma, squamous cell carcinoma and neuroendocrine carcinoma. Among the diverse histological types, gastric adenocarcinoma (GAC) is the most common GC. 2 In accordance with the degree of tumour tissue differentiation, GAC is further divided into highly differentiated, moderately differentiated, poorly differentiated adenocarcinoma, mucinous adenocarcinoma and signet ring cell carcinoma. Considerable amounts of clinical data have shown a higher 5-year survival rate in GC patients that present at an early stage compared with those patients that present at a late stage. 3 It has also been noted that GC patients with lymph node metastasis have a shorter postoperative survival time than those with an absence of lymph node metastasis. 4 Despite these clinical observations, the exact pathological mechanisms of GC remain unclear.
The vascular endothelial growth factor receptor (VEGFR) has been well recognized to have a pivotal role in the development of lymphatic system. 5 The ligands for VEGFR include vascular endothelial growth factor (VEGF)-A (the most extensively studied ligand known for its role in angiogenesis), VEGF-B, VEGF-C, VEGF-D and placental growth factor. 6 The interaction between VEGFR and its ligands is crucial for normal vascular development and function, as well as in pathological conditions such as cancer.7,8 VEGFR consists of seven immunoglobulin-like domains (i.e. transmembrane domain, ligand-binding domain, intracellular tyrosine kinase domain). 9 In mammals, the VEGFR family is composed of three membrane proteins: VEGFR-1, VEGFR-2 and VEGFR-3; of which, VEGFR-3 is primarily expressed in the endothelial cells of the lymph node, where it serves as a lymphatic endothelial marker in the vessels.10,11 Growing evidence has shown that VEGFR-3 is abnormally upregulated in a variety of malignant tumours,10,11 suggesting its involvement in lymphangiogenesis and tumour metastasis. VEGFR-3 is also expressed in neuronal progenitor cells, osteoblasts, macrophages and neural stem cells;12–15 and appears to participate in the development and progression of malignancies. 16 To date, the exact function of VEFGR-3 in GAC remains unclear and the molecular mechanisms need to be elucidated. Further insights into the molecular mechanisms for the potential roles of VEGFR-3 in malignant tumours, including GC, are expected to lay a strong scientific foundation to guide the development of new and effective approaches for the early diagnosis, prognostic evaluation and targeted therapy for cancer patients. The objective of this current study was to undertake in vivo and in vitro investigations designed to determine the pathological role of VEGFR-3 in GC and to explore the underlying molecular mechanisms.
Materials and methods
Study design
The expression levels of the VEGFR-3 gene in GC tissues were analysed using The Cancer Genome Atlas (TCGA) database. The VEGFR-3 gene expression profiles of 408 GC tissue samples, 36 histologically normal gastric specimens and clinical characteristics were acquired from TCGA database. Overall survival (OS) curves were plotted for GC patients to evaluate the potential relationship between VEGFR-3 gene expression and OS. Meanwhile, the effects of VEGFR3 on proliferation, migration, invasion and apoptosis of GC cell lines and the related molecular mechanisms were investigated by in vivo animal experiments and in vitro cytological function experiments. This study was based on publicly available source data obtained from TCGA database. Neither human tissue/samples nor personal information were used. Ethical committee approval was not required.
Cell culture
Five human GC cell lines (AGS, MKN-45, BGC-803, MGC-823, HGC-27) and one human normal gastric mucosal epithelial cell line (GES-1) were purchased from the cell bank of the Chinese Academy of Sciences (Shanghai Cancer Institute, Shanghai, China) or the American Type Culture Collection (Gaithersburg, MD, USA). GC cells and GES-1 cells were grown in RPMI-1640 medium supplemented with 10% fetal bovine serum (both from Thermo Scientific, Shanghai, China) and 1% penicillin-streptomycin (Beijing Solaibo Technology, Beijing, China) in an incubator at 37°C and 5% CO2.
Animals
BALB/c immunocompromised mice aged 4–6 weeks (weight range, 12–16 g) were provided by Beijing Vitong Lihua Experimental Animal Technology Co, Ltd. (Beijing, China) and the experimental animals were reared in specific pathogen-free facilities. Animals had free access to water and food. They were housed under a 12-h light/12-h dark cycle and were maintained in well-ventilated, clean conditions. The research protocols for experiments involving animals were reviewed and approved by Experimental Animal Ethics Committee of Weifang Medical College (No. 2021SDL318). The handling of experimental animals was in compliance with the Regulations on the Management of Experimental Animals. This study was conducted in compliance with the ARRIVE guidelines.
Mouse models of GC
Lentiviral vector carrying VEGFR-3 short hairpin RNA (shRNA) (LV-shVEGFR-3 group) or non-specific shRNA as a control (LV-NC group) (Gene Pharma, Shanghai, China) was introduced into human GC cells (MKN-45). A total of 5 × 105 MKN45 cells transfected with VEGFR-3 shRNA in the experimental group or non-specific shRNA in the control group were injected subcutaneously into the forelimb of nude mice (n = 5 each group) to construct a gastric tumour-bearing mouse model. Tumour growth, size and weight were recorded for the experimental and control groups. In addition, a mouse model of distant metastasis (metastases in the lung) was established by injecting MKN45 cells into the nude mice (n = 5 each group) through the caudal vein.
VEGFR-3 gene specific small interfering RNA transfection
VEGFR-3 gene specific small interfering RNA (siRNA) and non-specific control siRNA (NC) were designed and synthesized by Gene Pharma. To silence VEGFR-3 gene expression, GC cells were transfected with VEGFR-3 gene specific siRNA. In brief, GC cells were plated in a 6-well plate. After the cell density reached 1 × 106 cells/well, VEGFR-3 siRNA or NC was transfected into GC cells using X-treme Gene Transfection Reagent (Roche, Shanghai, China) following the manufacturer’s instructions. The silencing efficiency of the VEGFR-3 siRNA was determined by reverse transcriptase–polymerase chain reaction (RT–PCR) analysis. The required siRNA and shRNA sequences are shown in Table 1.
Small interfering (si) RNA and short hairpin (sh) RNA sequences used to knockdown vascular endothelial growth factor receptor-3 (VEGFR-3) gene expression.

VEGFR3, vascular endothelial growth factor receptor-3; NC, normal control.
EdU cell proliferation assay
The 5-ethynyl-2′-deoxyuridine (EdU) cell proliferation assay was used to measure cell proliferation. In brief, cells were seeded in a 96-well plate in triplicate for each group. Then 100 μl of 50 μM EdU was added and incubated at 37°C for 2–3 h. Upon completion, the cells were fixed in 4% paraformaldehyde, followed by incubation with 50 μl glycine (2 mg/ml) at room temperature for 5 min. After washing for 10 min at room temperature using 100 μl 0.01 M phosphate-buffered saline (PBS; pH 7.4) with 0.5% Triton™ X-100 (PBS-T), cells were incubated with 100 μl of 1× Apollo staining reaction solution in the dark for an additional 30 min. Upon completion, 30 μl 4′,6-diamidino-2-phenylindole (DAPI) was added and incubated in the dark for 5 min. After another washing with 0.01 M PBS (pH 7.4), cells were cultured for 30 min with 100 μl Hoechst 33342. The number of EdU-positive cells was counted under a BX51 fluorescence microscope (Olympus, Tokyo, Japan).
Cell cloning assay
In vitro clonogenic assay was performed 48 h after transfection to examine the cell proliferation capability. Briefly, cells were seeded in a 6-well plate at a density of 1000 cells per well and maintained in an incubator for 2–3 weeks. The cell culture was terminated when most cells formed clone balls of ≥50 cells. Subsequently, cells were fixed in 4% paraformaldehyde and 0.1% crystalline purple. Cloned spheres were examined under an inverted microscope.
Transwell cell migration and invasion assays
The cell migration and invasion assays were conducted using Corning® Transwell® cell culture inserts with a pore size of 8 µm (Corning Company, Corning, NY, USA). A total of 2 × 105 cells were inoculated in the upper chamber. After 48 h of incubation with 200 μl serum-free cell culture medium, the infiltrated cells were fixed with 4% paraformaldehyde. The cells were stained using 0.1% crystal violet solution. The number of cells passing through the chambers in three fields was randomly counted using a high magnification NIB410 microscope (Leica Microsystems Trading, Shanghai, China). During the cell invasion assay, cells were pretreated with matrix gel to measure the capability of cell invasion through the extracellular matrix. The procedures were similar to the transwell cell migration assay.
Cell scratch test
The cell scratch test was conducted to evaluate wound healing, a process involving cell migration as a critical step. In brief, cells were cultured in a 6-well plate for 24 h and then the monolayer of cells in each well was damaged using a 200 μl pipette tip. After removing the floating cells with 0.01 M PBS (pH 7.4), the cells were cultured for another 48 h. The wound healing process was visualized and analysed under an NE910 inverted light microscope (Leica Microsystems Trading).
Flow cytometry to measure apoptosis
The cells were collected, re-suspended in 400 μl 1× Binding Buffer and then incubated with 5 μl Annexin V-fluorescein isothiocyanate and propidium iodide at room temperature for 15 min. The apoptotic cells were detected by flow cytometry using an Attune NxT flow cytometer (Thermo Scientific).
Quantitative PCR
Quantitative PCR was performed to examine the mRNA levels of VEGRF-3. Total RNA was extracted from 8–9 × 105 cells using the TrizolTM RNA Extraction Kit (Invitrogen, Carlsbad, CA, USA) and the concentration was quantified with the RNeasy Mini Kit (Thermo Scientific). cDNA was synthesized by the cDNA Reverse Transcription Kit (Toyobo, Osaka, Japan). PCR was performed using a quantitative PCR kit (Roche). The relative mRNA level of VEGFR-3 was calculated using the 2−△△ Ct method. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal reference. The primers for quantitative PCR were provided by Shanghai Sangon Biotech (Shanghai, China) as follows: VEGFR3, forward 5ʹ-CCTGACCATCCACAACGTCA-3ʹ, reverse 5ʹ-CACAATGACCTCGGTGCTCT-3ʹ; GAPDH, forward 5ʹ-GAGTCAACGGATTTGGTCGT-3ʹ, reverse 5ʹ-GACAAGCTTCCCGTTCTCAG-3ʹ. The cycling programme included an initial denaturation at 95°C for 10 min, followed by 40 cycles of denaturation at 95°C for 15 s, annealing at 60°C for 1 s, and elongation at 95°C for 15 s, followed by a final elongation step at 60°C for 1 min.
Western blot analysis
The levels of proteins of interest were examined using Western blot analysis. Briefly, the cells were collected, followed by total protein extraction with RIPA lysis buffer. The total protein concentration in the cell extract was quantified with the BCA protein assay. Total protein samples were loaded on sodium dodecyl sulphate–polyacrylamide gel electrophoresis and separated by electrophoresis. After completion of the electrophoresis, the gel with separated proteins was immediately transferred to a polyvinylidene fluoride membrane. The membrane was then blocked with 5% skimmed milk for 2 h at room temperature. The resulting membrane was incubated with primary antibodies (all used at 1:500–1000 dilutions; rabbit antihuman VEGFR3, vimentin, Bcl2, Bax and Caspase-3 antibodies from Abcam®, Cambridge, MA, USA; and rabbit antihuman E-cadherin, N- cadherin and poly (ADP-ribose) polymerase [Parp] antibodies from Cell Signaling Technology®, Danvers, MA, USA) overnight at 4°C. The membrane was washed three times with 1 × Tris-buffered saline Tween-20 (TBST; pH 7.6) buffer and subsequently incubated with secondary horseradish peroxidase-conjugated antirabbit antibody (1:1000 dilution; Biyuntian Biotechnology Company of Shanghai, Shanghai, China) at room temperature for 1 h. The membrane was then washed three times with 1 × TBST (pH 7.6) buffer. Chemiluminescence (Shanghai Qinxiang Scientific Instrument Company, Shanghai, China) was used to develop the protein bands. The levels of protein were quantitatively analysed by ImageJ software (open access software, developed by the National Institutes of Health, USA.
Haematoxylin & eosin and immunohistochemistry staining
Gastric tumour tissues were fixed and embedded in paraffin. The resulting tissue block was cut into a 4-μm sections for haematoxylin and eosin (H&E) staining as follows. The slides were dewaxed and stained with haematoxylin for 3 min, then rinsed with distilled water for 5 min and differentiated with 1% hydrochloric acid alcohol for 10 s, followed by washing in distilled water for 1 min. The staining was returned to blue with 1% ammonia water for 5–10 s, then rinsed with distilled water for 1 min. The slides were then stained with eosin (water-soluble) for 1 min followed by washing in distilled water for 20 s.
Other selected wax blocks were cut into 4-μm sections and prepared for immunohistochemical staining as follows. After washing three times in 0.01 M PBS (pH 7.4) for 5 min each time at room temperature, the sections were incubated with an appropriate amount of hydrogen peroxide at room temperature for 10 min. The sections were washed three times with 0.01 M PBS (pH 7.4) for 5 min each time at room temperature. The sections were then incubated with undiluted primary rabbit antihuman antibodies to Ki-67 and N-cadherin (Maixin, Fuzhou, China) at 4°C overnight. The sections were washed three times with 0.01 M PBS (pH 7.4) for 5 min each time at room temperature. The sections were then incubated with undiluted horseradish peroxidase goat antirabbit secondary antibody (Maixin) at 37°C for 60 min, followed by washing three times with 0.01 M PBS (pH 7.4) for 5 min each time at room temperature. The sections were then incubated with a 3,3′-diaminobenzidine colour kit (Beijing Zhongshan Jinqiao Biotechnology, Beijing, China) and the nuclei were counterstained with haematoxylin. The criteria for interpretation of the immunohistochemical results were as follows. The detection of yellow and/or brownish-yellow substances in the nucleus or cytoplasm of the target cell was considered positive under light microscopy, excluding non-specific staining and/or colouration. Based on positive staining in the nucleus or cytoplasm, the results were categorized as follows: (i) <10% positive cells was considered negative (–); 10–25% positive cells was weakly positive (+); 26–50% positive cells was moderately positive (++); and >50% positive cells was strongly positive (+++). To simplify the calculations, positive expression was considered to include + to +++.
Statistical analyses
All statistical analyses were conducted using GraphPad Prism 9.0 software (GraphPad Software, San Diego, CA, USA). When comparing the means of three groups, if data were normally distributed, an analysis of variance (ANOVA) along with a post-hoc Tukey test was used to analyse the difference in means between the groups; if data were not normally distributed, the Kruskal–Wallis test (a non-parametric test) was used, after which the Dunn's test (a non-parametric test) was used for pairwise comparison between each independent group. When comparing the means of two groups, when data followed a normal distribution, a Student's t-test was used; when data did not follow a normal distribution, the Mann–Whitney U-test (a non-parametric test) was used. When comparing the means of tumour volumes in different groups at various time-points, a two-way ANOVA was used, which was followed by the Bonferroni method for pairwise comparison between each independent group or the Tukey method for comparison of different time-points within each group. The Kaplan–Meier method was used for the survival analysis; and the Log-rank (Mantel–Cox) test was conducted to examine the difference in means between the two groups. A P-value of <0.05 was considered to be statistically significant.
Results
VEGFR-3 gene expression is elevated in GC and high expression is related to a poor prognosis
The VEGFR-3 gene expression profiles of 408 GC tissues and 36 histologically normal gastric specimens as the controls were investigated. The VEGFR-3 gene expression levels in GC tissues were significantly greater than those in the histologically normal gastric specimens (P < 0.05) (Figure 1a). The relationship between levels of VEGFR-3 and the clinicopathological characteristics (e.g. lymph node metastasis, clinical and pathological stages) were assessed and the findings demonstrated a significant relationship between the levels of VEGFR-3 gene expression and the pathological stage (P < 0.05) (Figure 1b) or the numbers of positive lymph nodes in GC patients (P < 0.05) (Figure 1c). Analysis of the survival curves revealed that GC patients with higher VEGFR-3 gene expression levels displayed a significantly poorer prognosis than those patients who had lower VEGFR-3 gene expression levels (Figure 1d) (P = 0.02).
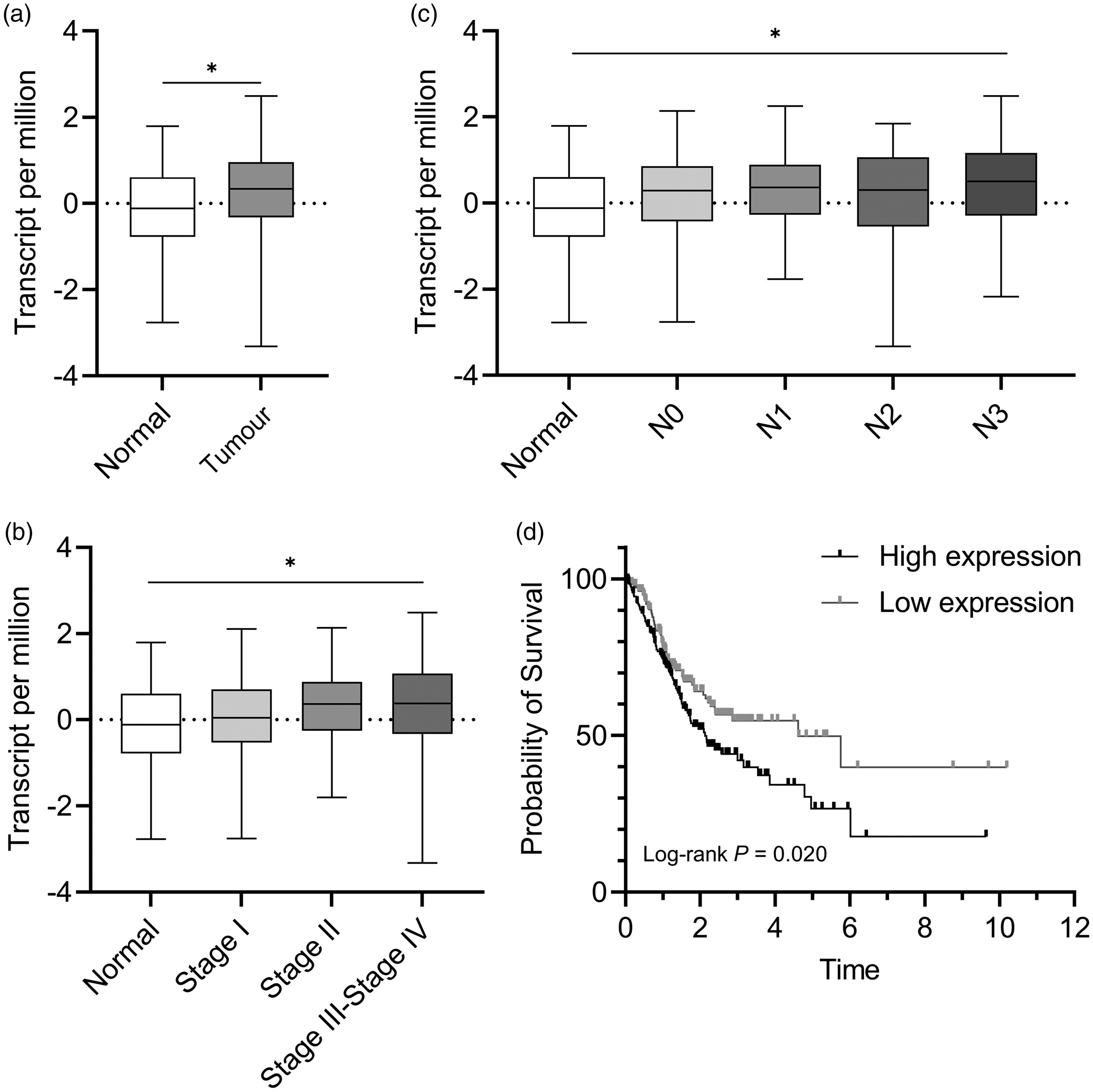
Vascular endothelial growth factor receptor-3 (VEGFR-3) gene expression and its association with prognosis in patients with gastric cancer (GC). The VEGFR-3 gene expression profiles of 408 GC tissue samples, 36 histologically normal gastric specimens and clinical characteristics were obtained from The Cancer Genome Atlas database: (a) expression levels of the VEGFR-3 gene in GC tissues and histologically normal gastric specimens; (b) expression levels of the VEGFR-3 gene in GC patients with different pathological stages; (c) expression levels of the VEGFR-3 gene in GC patients with different numbers of metastatic lymph nodes and (d) survival curve analysis of the relationship between VEGFR-3 gene expression levels and the prognosis of patients with GC. Box-whisker plots in a, b and c: black central line is the median; extremities of the box are the 25th and 75th percentiles; and the error bars are the minimum and maximum outliers; *P < 0.05, one-way analysis of variance.
Consistent with increased VEGFR-3 gene expression levels in patients with GC, Western blot analysis demonstrated significantly higher VEGFR-3 protein levels in GC cell lines compared with human immortalized non-tumour gastric mucosa cells (see supplementary materials, Figure 1) (MKN45, P < 0.0001; BGC-823, P = 0.0003). Among the five GC cell lines, MKN45 and BGC823 appeared to have higher levels of VEGFR-3 protein compared with the normal gastric mucosal epithelial cell line GES-1, thus, they were selected for subsequent cell culture experiments.
Knockdown of VEGFR-3 gene expression suppresses the proliferation and invasion of GC cells, and enhances apoptosis
To assess the biological roles of VEGFR-3 protein, VEGFR-3 siRNA was used to silence the gene expression in GC cells, and cellular processes associated with cancer growth, metastasis, and progression were examined. As expected, the transfection of GC cells with two VEGFR-3 siRNAs (si-VEGFR3-1/si-VEGFR3-2) significantly reduced VEGFR-3 mRNA levels in MKN45 and BGC823 cells (see supplementary materials, Figure 2) (P < 0.001 for all comparisons). EdU cell proliferation assays showed that silencing of the VEGFR-3 gene with si-VEGFR3-1/si-VEGFR3-2 led to a significant reduction of the proportion of positive proliferating MKN45 and BGC823 cells (Figure 2) (MKN45 si-VEGFR3-1, P = 0.0249; MKN45 si-VEGFR3-2, P = 0.0138; BGC823 si-VEGFR3-1, P = 0.0016; BGC823 si-VEGFR3-2, P = 0.003). The results of the cell cloning assays showed a significantly lower number and volume of clonal spheres formed by MKN45 and BGC823 GC cells transfected with VEGFR-3 siRNAs compared with the NC group (Figure 3) (MKN45 si-VEGFR3-1, P = 0.0015; MKN45 si-VEGFR3-2, P = 0.0015; BGC823 si-VEGFR3-1, P = 0.0025; BGC823 si-VEGFR3-2, P = 0.0015). Transwell cell migration and invasion assays showed that MKN45 and BGC823 cells transfected with VEGFR-3 siRNAs significantly inhibited the migration and invasion of GC cells (Figures 4a–4c) ([A] MKN45 si-VEGFR3-1 P = 0.0001, si-VEGFR3-2 P < 0.0001; BGC823 si-VEGFR3-1 P = 0.0004, si-VEGFR3-2 P = 0.0006; [B] MKN45 si-VEGFR3-1 P < 0.0001, si-VEGFR3-2 P < 0.0001; BGC823 si-VEGFR3-1 P < 0.0001, si-VEGFR3-2 P < 0.0001; [C] MKN45 si-VEGFR3-1 P = 0.0019, si-VEGFR3-2 P = 0.0008; BGC823 si-VEGFR3-1 P < 0.0001, si-VEGFR3-2 P = 0.0002). Furthermore, the apoptotic rate in the VEGFR-3 siRNA groups were significantly enhanced compared with the NC group, suggesting that silencing of VEGFR-3 gene expression significantly promoted apoptosis (P < 0.001) (Figure 5).

The 5-ethynyl-2′-deoxyuridine (EdU) cell proliferation assay was used to measure the effects of vascular endothelial growth factor receptor-3 (VEGFR-3) gene silencing on the proliferation of gastric cancer (GC) cells. GC cells (MKN45, BGC823) were transfected with two VEGFR-3 gene specific small interfering RNA (siRNA) or non-specific siRNA as the control (NC). EdU assays were performed to examine the effects of silencing VEGFR-3 gene expression on the number of proliferating cells: (a) fluorescent images from the EdU assays for the effects of transfected siRNAs on the proliferation of MKN45 and BGC823 cells (scale bar, 20 μm) and (b) quantitative analysis of the effects of transfected siRNAs on the proliferation of MKN45 and BGC823 cells. The horizontal black lines are the medians. DAPI, 4′,6-diamidino-2-phenylindole. *P < 0.05, **P < 0.01, one-way analysis of variance. The colour version of this figure is available at: http://imr.sagepub.com.

Cell cloning assays of the effects of vascular endothelial growth factor receptor-3 (VEGFR-3) gene silencing small interfering (si)RNAs on the proliferation of gastric cancer (GC) cells. MKN45 and BGC823 cells were transfected with two VEGFR-3 gene specific siRNAs or non-specific siRNA as the control (NC). Cell cloning assays were conducted to determine the effects of VEGFR-3 siRNAs on the proliferation of GC cells. The upper panels display the representative images of the cell cloning assays for the effects of transfected siRNAs on the proliferation of MKN45 and BGC823 cells (scale bar, 10 μm). The lower bar graphs show the quantitative analyses of the effects of VEGFR-3 siRNAs on the proliferation of MKN45 and BGC823 GC lines. The horizontal black lines are the medians. **P < 0.01, one-way analysis of variance. The colour version of this figure is available at: http://imr.sagepub.com.

Inhibitory effects of vascular endothelial growth factor receptor-3 (VEGFR-3) gene silencing small interfering RNA (siRNA) on the migration and invasion of gastric cancer (GC) cells. MKN45 and BGC823 cells were transfected with two VEGFR-3 gene specific siRNAs or non-specific siRNA as the control (NC). Transwell migration and invasion assays were performed: (A) cell scratch assays of the effects of transfected VEGFR-3 siRNAs on the migration ability of GC cells (scale bar, 10 μm); transwell assays of the effects of transfected VEGFR-3 siRNAs on the (B) migration and (C) invasion of GC cells (scale bar, 20 μm). The horizontal black lines are the medians. **P < 0.01, ***P < 0.001, one-way analysis of variance. The colour version of this figure is available at: http://imr.sagepub.com.

Effects of vascular endothelial growth factor receptor-3 (VEGFR-3) gene silencing small interfering RNA (siRNA) on the apoptosis of gastric cancer (GC) cells. MKN45 and BGC823 cells were transfected with two VEGFR-3 gene specific siRNAs or non-specific siRNA as the control (NC). Flow cytometry was carried out to determine the proportion of apoptotic cells. The apoptotic cell rate was significantly higher in MKN45 and BGC823 cells transfected with VEGFR-3 siRNAs compared with the NC group. The horizontal black lines are the medians. ***P < 0.001 versus the NC group; one-way analysis of variance. The colour version of this figure is available at: http://imr.sagepub.com.
Given that E-cadherin exerts an inhibitory role as a suppressor of tumour growth and metastasis in epithelial cancers, the current study examined the effects of VEGFR-3 siRNAs on the protein levels of E-cadherin and N-cadherin in GC cells. Western blot analysis demonstrated that silencing of the VEGFR-3 gene significantly enhanced E-cadherin protein levels, but led to significant decreases in N-cadherin and vimentin protein levels in MKN45 and BGC823 cells (P < 0.05 for all comparisons) (see supplementary materials, Figure 3), thereby inhibiting the epithelial–mesenchymal transition (EMT). Consistent with these findings, the knockdown of the VEGFR-3 gene significantly suppressed B-cell lymphoma 2 (Bcl2) in BGC823 and MKN45 GC cells, but significantly increased Bcl2-associated X protein (BAX), caspase 3 and Parp proteins. The Bcl2, BAX, caspase 3 and Parp protein levels differed significantly between the VEGFR-3 siRNA and NC groups (P < 0.05 for all comparisons) (see supplementary materials, Figure 4).
Silencing of the VEGFR3 gene inhibits tumour growth and metastasis in mouse models of GC and lung metastasis
Based on the in vitro findings, the current study then determined the effects of VEGFR-3 gene expression on in vivo tumour growth and metastasis using mouse models of GC and distant metastasis in the lung. The knockdown of VEGFR-3 gene expression inhibited tumour growth in the LV-shVEGFR-3 group compared with the LV-NC group (Figure 6a). VEGFR-3 shRNA-mediated inhibitory effects on gastric tumour growth were further supported by evidence showing significant reductions in both tumour volume and weight in the LV-shVEGFR-3 group compared with the LV-NC control group (Figures 6b and 6c) ([B] LV-NC week 3 P = 0.0113, week 4 P = 0.0010, week 5 P < 0.0001; [C] P = 0.0097). The data from H&E and IHC staining of subcutaneous tumour tissues showed that the ability to invade the capsule was diminished; and the Ki-67 index in the gastric tumour tissues was reduced in the LV-shVEGFR-3 group compared with the LV-NC group (Figures 6d and 6e).
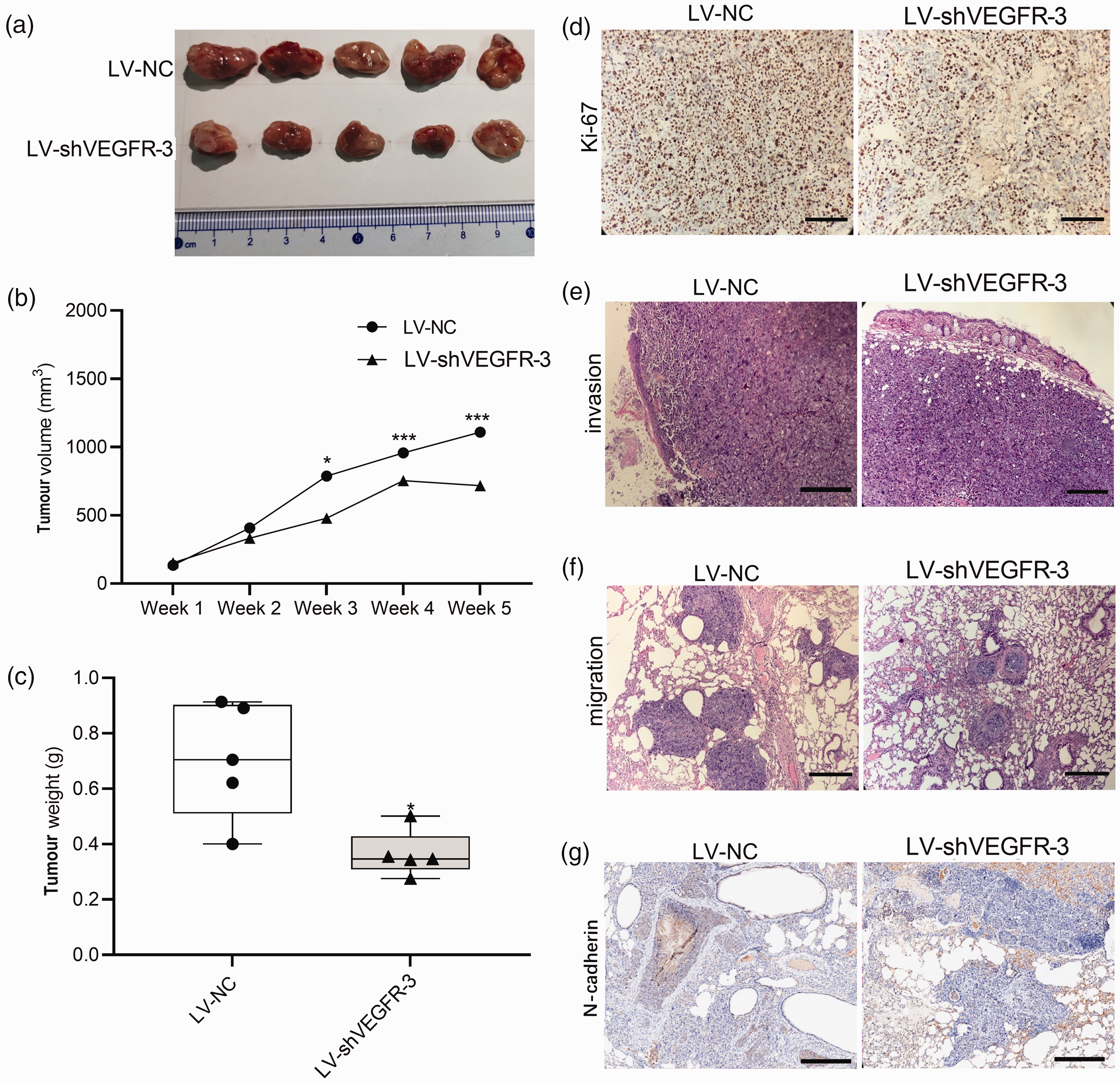
The in vivo effects of altering the vascular endothelial growth factor receptor-3 (VEGFR-3) gene expression on tumour growth and metastasis in mouse models of gastric cancer (GC) and distant metastasis in the lung. The mice were treated with lentiviral vector carrying VEGFR-3 short hairpin RNA (shRNA) (LV-shVEGFR-3 group) or non-specific shRNA as a control (LV-NC group): (a) effects of VEGFR-3 gene knockdown on tumour growth; (b) effects of VEGFR-3 gene knockdown on tumour volume; (c) effects of VEGFR-3 knockdown on tumour weight; (d) effects of VEGFR-3 gene knockdown on the levels of Ki-67 protein in subcutaneous tumour as detected by immunohistochemistry (scale bar, 20 μm); (e) effects of VEGFR-3 gene knockdown on invasion as detected by haematoxylin & eosin staining (scale bar, 20 μm); (f) effects of VEGFR-3 gene knockdown on the migration ability as observed in the caudal vein (haematoxylin & eosin staining; scale bar, 20 μm) and (g) effects of VEGFR-3 gene knockdown on the expression of N-cadherin in subcutaneous tumour as detected by immunohistochemistry (scale bar, 20 μm). Box-whisker plots in c: black central line is the median; extremities of the box are the 25th and 75th percentiles; and the error bars are the minimum and maximum outliers; *P < 0.05, ***P < 0.001, one-way analysis of variance. The colour version of this figure is available at: http://imr.sagepub.com.
The in vivo investigations on lung metastasis showed that silencing of the VEGFR-3 gene expression reduced the number of lung metastases in the LV-shVEGFR-3 group compared with the LV-NC group (Figure 6f). In addition, the diameter of the metastases was smaller in the LV-shVEGFR-3 group compared with the LV-NC control group (Figure 6f). The resulting data in the mouse models of GC and lung metastasis indicated that knockdown of VEGFR-3 gene expression inhibited tumour growth, invasion and metastasis. To investigate the potential involvement of the EMT pathway in the effects of VEGFR-3 gene expression on tumour invasion ability, the levels of N-cadherin protein in lung metastases were examined. IHC analysis revealed a decrease in the levels of N-cadherin in the LV-shVEGFR-3 group compared with the LV-NC control group (Figure 6g).
Discussion
The results of the present study revealed the following novel findings: (i) VEGFR-3 protein levels were significantly increased in GAC and its higher levels were related to a poorer prognosis; (ii) silencing of VEGFR-3 gene expression significantly diminished the GC cell growth and invasion, but induced cell apoptosis; (iii) in vivo investigations indicated that the knockdown of VEGFR-3 gene expression significantly suppressed the growth of GC in mice; (iv) in the mouse model of lung metastasis, silencing of VEGFR-3 gene expression significantly decreased the number and size of metastatic tumours; (v) mechanistic studies suggested that VEGFR-3 is directly involved in the downregulation of vimentin, N-cadherin and Bcl-2, but in the upregulation of BAX, PARP, E-cadherin and caspase 3 in GC cells. Together, these findings indicate that VEGFR-3 directly affects key molecules in the apoptotic and EMT signalling pathways, exerting its pathological role via enhancing the GC growth and metastasis.
Vascular endothelial growth factor receptor is primarily identified as a lymphatic growth factor receptor specific to lymphatic endothelial cells. Increased VEGFR-3 expression is associated with poor prognosis of patients with various malignant tumours.17,18 A previous study demonstrated that increased levels of VEGFC and VEGFR-3 were related to metastasis in the lymph nodes. 19 Another study reported that VEGFR-3-positive lymphatic vessel density was significantly elevated in GC and its higher levels were associated with the size of the primary tumour, lymphatic infiltration and metastasis in the lymph nodes. 20 In our previous study, VEGFR-3 was elevated in early GC and the higher levels were related to the depth of tumour invasion in the lymph nodes. 21 A previous meta-analysis found that greater levels of VEGFR-3 were associated with the higher invasion and metastasis of GC. 22 In this current study, the TCGA database GAC cohort analysis revealed that VEGFR-3 gene expression was highly upregulated in GAC, which was associated with a poorer prognosis of GAC patients and lymph node metastasis.
The current study used siRNA to silence the gene expression of VEGFR-3 to elucidate the role of VEGFR-3 in GC. The results obtained from in vitro cytology experiments indicated that the knockdown of VEGFR-3 gene expression diminished the GC cell growth, migration and invasion, while inducing GC cell apoptosis. A previous study demonstrated that the downregulation of VEGFR-3 protein levels by VEGFR-3 siRNA in colon cancer cells promoted apoptosis and inhibited the invasion of colon cancer cells. 23 Inhibition of VEGFR-3 significantly inhibited the growth of lung adenocarcinoma A549 cells, as well as cell migration and invasion in the tumour microenvironment. 24 To better understand the roles of VEGFR-3 gene expression in GC cell growth, invasion and migration, in vivo investigations were conducted in mouse models of GC and lung metastasis. The results of subcutaneous tumorigenesis and caudal venous injection confirmed that the silencing of VEGFR-3 gene expression demonstrated an inhibitory effect on GC cell growth, migration and invasion. These results suggested that VEGFR-3 has a promoting effect on GC cell growth, invasion and migration, but an inhibitory effect on the GC cell apoptosis.
A number of previous studies have reported that new lymphatic vessel formation (also referred to as lymphangiogenesis) within tumour tissues is achieved mainly via regulating the levels of VEGFR-3 in lymphatic endothelial cells.17,25,26 It has been shown that VEGFR-3 activates the protein kinase B (PKB/Akt) and mitogen-activated protein kinase (MAPK) pathways, thereby promoting cell proliferation and migration.17,25,26 The VEGF-C-VEGFR3/FLT4 axis can modulate the growth and metastasis of breast cancer in an autocrine manner. 18 Currently, mechanistic studies of VEGFR-3 have been performed with the main focus on the VEGF-C/VEGFR-3 signalling axis.27–29 Neuropilin2 can bind to VEGF-C/D and interact with VEGFR-3 to form a complex, thereby leading to activation of VEGFR-3 signalling and promoting lymphatic endothelial cell proliferation and lymphangiogenesis. 29 The VEGFC/VEGFR3 signalling pathway regulates the proliferation of mouse spermatogonia through activating the cyclin D1 and Akt/MAPK signalling pathways, and modulates cell apoptosis via Bcl-2 and caspase 3/9. 30 Expression of the VEGF-C/VEGFR3 axis in the KRAS/YAP1/Slug pathway induces the migration, invasion and desiccation of skin cancer cells. 30 A previous study found that the VEGFC/VEGFR-3 axis mediates transforming growth factor inhibitor 1 to induce the EMT of non-small cell lung cancer cells. 31 Another study reported that the VEGF-C/PI3K/Akt signalling pathway exerted an inhibitory effect on GC cell growth and invasion. 32 Recently, it has been shown that tumour-associated macrophages induce both VEGFR-3 and VEGF-C gene expression in lung adenocarcinoma cells, 24 leading to enhancement of the migration and invasion capacity. Interestingly, blocking VEGFR-3 signalling results in inhibitory effects on tumour growth by upregulating the expression of p53 and phosphatase and tensin homolog. 24 In addition, blocking the VEGFR-3-mediated signal transduction pathway has been reported to effectively suppress tumour lymphangiogenesis. 33
Based on the existing findings of the mechanisms for the VEGF-C/VEGFR-3 signalling axis in the cell growth, apoptosis and invasion, the current study determined whether VEGFR-3 alone could regulate GC cell proliferation, apoptosis and invasion. The knockdown of VEGFR-3 gene expression suppressed GC cell migration and invasion. Given that the EMT promotes the tumour cell migration via impairing the adhesive ability of tumour cells, thereby inducing tumour metastasis, the current study hypothesized that VEGFR-3 could affect GC cell migration and invasion ability by regulating the EMT. Consistent with this hypothesis, the current study subsequently found that silencing VEGFR-3 gene expression altered the expression of key EMT genes (i.e. N-cadherin, vimentin, E-cadherin) in GC cells. Specially, knockdown of VEGFR-3 gene expression in GC cells increased E-cadherin (an epithelial marker) as well as decreasing N-cadherin and vimentin (interstitial markers). Additionally, it was found that the levels of N-cadherin were reduced after the knockdown of VEGFR-3 gene expression in lung metastasis. These findings suggest that VEGFR-3 might exert its promotive effects on GC cell migration and invasion via regulating the EMT pathway.
It is also worth noting that the knockdown of VEGFR-3 gene expression induced the apoptosis of GC cells. However, the mechanism responsible for the regulation of GC cell apoptosis by VEGFR-3 remained unclear. Therefore, the current study analysed the key genes known to be involved in apoptotic pathways, such as BAX, caspase 3, Bcl-2 and PARP in GC cells using siRNA. The results demonstrated the downregulation of Bcl-2 in response to the knockdown of VEGFR-3 gene expression, and the upregulation of BAX, PARP and caspase 3. Therefore, these current findings suggest that VEGFR-3 affects the apoptosis of GC cells through regulating the endogenous apoptotic pathways mediated by caspase 3 and Bcl-2/BAX.
This study has several limitations. First, the analysis of VEGFR3 gene expression in GC and its relationship with clinicopathology was based on data from the TCGA database. Therefore, future studies should validate these findings using a large cohort of patients with GC. Secondly, the molecular mechanisms and signalling pathways underlying the effects of VEGFR3 on the proliferation, migration, invasion and apoptosis of GC cells need further investigation in the future.
In conclusion, this current study demonstrated that VEGFR-3 induces the growth and metastasis of GC by directly regulating key molecules in the apoptotic and EMT signalling pathways. As such, the present study may improve understanding of the pathological role of VEGFR-3 in GC. The molecular pathways involved in the VEGFR-3-mediated promotion of cancer growth and metastasis hold potential as targets in developing novel approaches to improve the diagnosis, prognosis and treatment of GC.
Supplemental Material
sj-pdf-1-imr-10.1177_03000605241234558 - Supplemental material for The pathological roles and potential mechanisms of vascular endothelial growth factor receptor-3 in gastric cancer
Supplemental material, sj-pdf-1-imr-10.1177_03000605241234558 for The pathological roles and potential mechanisms of vascular endothelial growth factor receptor-3 in gastric cancer by Xiu-Feng Li, Xiu-Juan Zhang, Fu-Rong Hao, Xiao-Tong Dong, Guo-Dong Xu and Yun-Xiang Zhang in Journal of International Medical Research
Footnotes
Author contributions
X.F.L. and Y.X.Z. participated in the design; Y.X.Z. and F.R.H. supplied administrative support; G.D.X., X.T.D. and X.F.L. conducted the experiment and collected and analysed the data; X.F.L. and X.J.Z. supplied critical reagents or animals; X.F.L. and X.T.D. wrote the manuscript. All authors participated in the review of the manuscript. X.F.L. and Y.X.Z contributed equally to this work.
Declaration of conflicting interest
The authors declare that there are no conflicts of interest.
Funding
This work was supported by the Scientific Research Project of Weifang Health and Family Planning Commission (Project No. 2017WSJS133) and Shandong Provincial Natural Science Foundation Project (Project No. ZR2021MH261).
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.