
Abstract
Caroli disease is a rare congenital malformation that predisposes to segmental cystic dilatation of the intrahepatic bile ducts. Banti syndrome is characterized by persistent splenomegaly due to chronic congestion, resulting in a low hematocrit and ultimately leading to pancytopenia. In this report, we describe a 29-year-old woman who presented with a >20-year history of hepatitis B surface antigen positivity and a >1-year history of recurrent fatigue and malaise. On examination, the patient had abdominal distension with marked splenomegaly (7 cm below the ribs) and ascites with tenderness of the abdominal muscles to palpation. A complete blood count showed a low white blood cell count, red blood cell count, and hemoglobin concentration. During the course of treatment, the patient developed multiple symptoms of pancytopenia and concomitant splenomegaly, and she was discharged after total splenectomy with good recovery. The combination of Banti syndrome and Caroli disease results in severe symptoms of portal hypertension.
Introduction
Caroli disease is a rare congenital malformation of the intrahepatic bile ducts, with a prevalence of less than 1 per million individuals and a male:female ratio of 1.0:1.8. 1 It is characterized by segmental cystic dilatation of the intrahepatic bile ducts and may be associated with biliary calculi, cholangitis, and liver abscesses. 2 In most cases, it is transmitted in an autosomal recessive manner; however, the mode of inheritance is still being explored. 3 Caroli disease is known as Caroli syndrome or Grumbach disease when associated with congenital liver fibrosis. Common symptoms of the disease include abdominal pain, fever, hyperbilirubinemia, elevated alkaline phosphatase, hepatomegaly, or signs of portal hypertension, and it is easily diagnosed in patients in their early 20s. 4 Common complications include intrahepatic stones, cholangitis, liver abscesses, pancreatitis, cholangiocarcinoma (7%), variceal hemorrhage, and ascites. 5
First described in the late 19th century by the Italian professor Guido Banti, Banti syndrome is also widely referred to as idiopathic portal hypertension and hepatic portal sclerosis. 6 Symptoms of Banti syndrome include an enlarged spleen and a reduced whole blood cell count due to hypersplenism and portal hypertension; however Banti syndrome does not affect the liver. The prevalence of this syndrome is low worldwide; overall, it is more common in developing countries and middle-aged people. 7
When Caroli disease is combined with Banti syndrome, the spleen is primarily affected. Other features include varicose veins detected on endoscopy, cytopenia in one or more cell lines, portal hypertension with significant portal shunting, and hepatomegaly. 8 Ruptured varices and life-threatening hemorrhage can easily complicate the presence of both syndromes. We herein describe a 29-year-old woman who presented with a 1-year history of recurrent weakness, abdominal pain, and abdominal distension, and strong tea-colored urine.
Case presentation
A 29-year-old woman who had been found to be hepatitis B surface antigen (HBsAg)-positive 20 years previously presented with abdominal pain and distension and weakness of the extremities. She had no history of jaundice, diarrhea, cough, abnormal sensation, chest pain, palpitations, dyspnea, itchy skin, or bleeding gums.
On general examination, the patient was thin, had a countenance consistent with chronic liver disease, and was supine in bed. She demonstrated good orientation to time, place, and person. Her body temperature was normal (36.11°C). Her pulse rate was 85 beats/minute and regular, exhibiting a normal pulse volume and other characteristics. The patient’s blood pressure was 130/68 mmHg. She had no jaundice, clubbing and cyanosis of fingers, or lymph node enlargement. Abdominal examination revealed an enlarged spleen, which was palpable 7 cm below the costal margin. The liver was also palpable. The abdomen was distended and negative for fluid shifts. The abdominal muscles were soft, and no pressure or rebound pain, dilated veins, or significant pulsations were detected. The rest of the systemic examination findings were normal. Liver function tests revealed an elevated direct bilirubin concentration, and renal function tests revealed an elevated uric acid concentration. The prothrombin time and international normalized ratio were high, and the prothrombin activity was low. Routine urinalysis was normal. The patient was positive for HBsAg and negative for anti-human immunodeficiency virus antibody and anti-hepatitis C virus antibody. Inflammatory markers showed slight abnormalities.
A complete blood count showed a white blood cell count of 2.17 × 109/L, red blood cell count of 2.24 × 1012/L, hemoglobin concentration of 70.00 g/L, mean red blood cell volume of 96.20, and platelet count of 76.00 × 109/L. The differential leukocyte count revealed 28.20% lymphocytes, 65.60% neutrophils, 1.50% eosinophils, 54.10% monocytes, and 0.60% basophils. Peripheral smears showed no malarial parasites. Abdominal computed tomography showed enlargement of the liver and spleen, and the spleen exhibited abnormal echoes. Pathologic examination showed dilated intrahepatic bile ducts with hepatic fibrosis consistent with Caroli disease, chronic fibrosclerotic splenomegaly, and vasodilatation and siltation in the fibrous connective tissue (Figures 1–4).
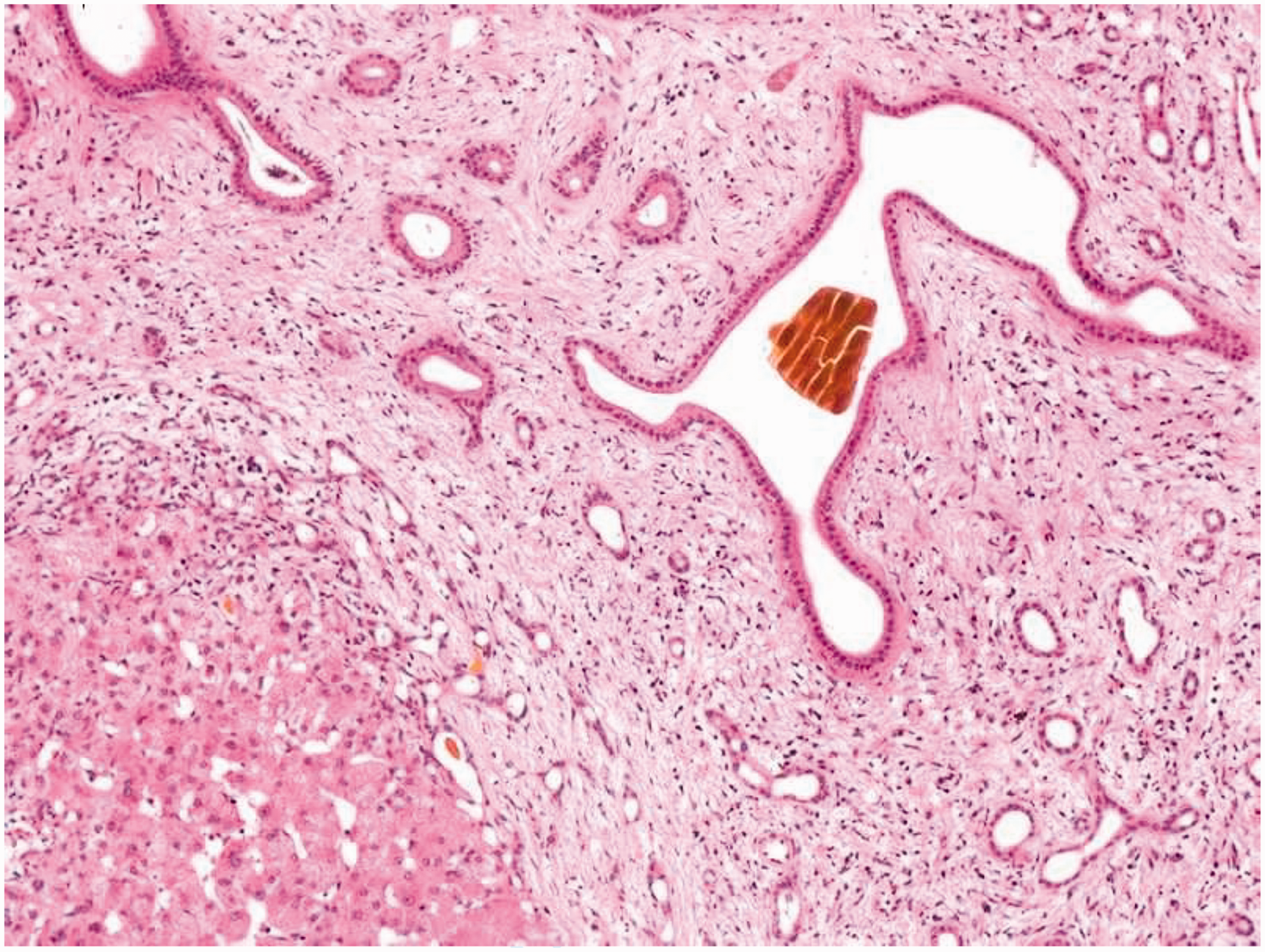
Structural disorganization of the hepatic lobules with irregular hepatocellular nodule formation. Examination revealed mild hepatocellular enlargement, dilated hepatic sinusoids around the central vein, interstitial fibrous tissue proliferation, widened fibrous septa, significant dilatation of the bile ducts, bile duct hyperplasia, bile thrombus formation, and peripheral diffuse inflammatory cell infiltration.

Special tests. Reticulostaining and Masson staining showed widened fibrous septa dividing the liver parenchyma, positive iron staining, positive periodic acid–Schiff staining, and negative diastase periodic acid–Schiff staining. Immunohistochemistry revealed negativity for hepatitis B surface antigen and hepatitis B core antigen and positivity for cytokeratin 7, cytokeratin 19, CD34, and pleural carcinoembryonic antigen.

Pathologic examination findings. Examination revealed fibrotic thickening of the splenic tegument, atrophy and reduction of splenic microsomes, dilatation and bruising of the hepatic sinusoids, hyperplasia and swelling of endothelial cells, and fibrotic hemorrhage around the splenic artery.
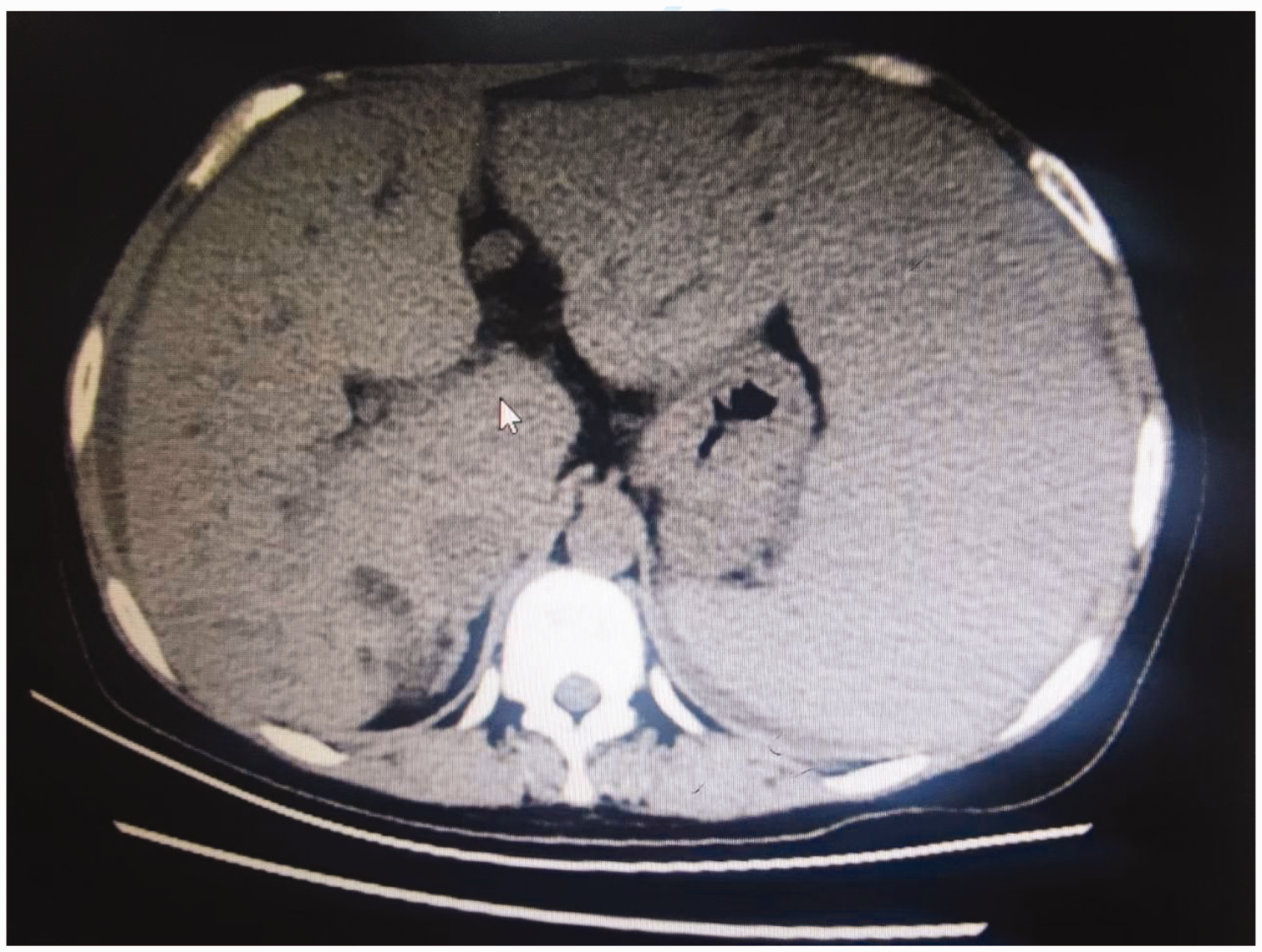
Abdominal computed tomography. The axial view without contrast showed an enlarged liver with no significant foci of abnormal intrahepatic enhancement. The spleen was enlarged and homogeneous and contained abnormal echoes within it.
Discussion
Caroli disease is a unique congenital malformation that results in segmental cystic dilatation of the intrahepatic bile ducts. Common symptoms include abdominal pain, fever, hepatomegaly, and portal hypertension. Banti syndrome usually results in chronic splenic congestion and enlargement, leading to destruction of blood cells. Over time, patients tend to vomit blood and develop black stools, causing the anemia to become more severe. Hepatic cirrhosis eventually develops, but splenomegaly remains the main symptom and is often accompanied by severe ascites. Symptoms of Banti syndrome include weakness, fatigue, anemia, and splenomegaly. Various factors predispose to Banti syndrome, including infectious factors (hepatitis B virus), coagulation abnormalities, and toxic metal exposure. The main symptom in our patient with Banti syndrome was splenomegaly, which we found during physical examination. In patients with portal hypertension, beta-blockers should be given for primary prophylaxis even in the absence of hepatic cirrhosis. When the two syndromes coexist, the patient's anemia worsens over time. Cirrhosis eventually develops, but the main symptom is splenomegaly along with possible ascites. Caroli disease combined with Banti syndrome has been reported in very few patients. Such patients frequently present with symptoms associated with a reduction in the three blood cell lineages, increasing the risk of severe infections and anemia. The etiology is multifactorial and may involve infectious agents (hepatitis B virus), coagulation abnormalities, and genetic factors. 9
There is some evidence to support immune abnormalities in patients with these syndromes.10,11 Tumor necrosis factor alpha (TNF-α) is involved in the maintenance and homeostasis of the immune system, inflammation, and host defense, and there is evidence that an increase in soluble TNF receptors I and II may cause portal hypertension. 12 In addition, because TNF activates the immune response, increases in interleukins 2, 4, 6, and 10 and interferon gamma are also observed in these patients.
The main symptoms in patients with Banti syndrome combined with Caroli disease are portal hypertension and splenomegaly, which were found in our case. In emergency situations, these conditions can be treated with endoscopic therapies (e.g., sclerotherapy or rubber band ligation) to reduce the risk of bleeding by closing the variceal channels. Even in the absence of cirrhosis, patients should receive vasoconstrictors that reduce portal inflow (terlipressin, growth inhibitors, or their analogues for acute hemorrhage and nonselective beta-blockers for prevention of initial bleeding and rebleeding) and vasodilators (e.g., isosorbide) that reduce portal pressure by decreasing intrahepatic resistance. 13 Splenectomy is indicated in patients with recurrent bleeding.
Conclusions
After our patient underwent total splenectomy and peripancreatic vascular dissection, the symptoms of portal hypertension were relieved, the complete blood cell count returned to normal, and the general clinical condition stabilized. Six months after the patient was discharged from the hospital, follow-up indicated good recovery and no complications. Banti syndrome causes slow-onset splenomegaly and blood cell destruction, which can lead to a decrease in all blood cell counts. When Banti syndrome is combined with Caroli disease, the patient develops severe symptoms of portal hypertension. Understanding this rare condition allows us to consider the best diagnostic approach, including the various differential diagnoses, and to begin the most appropriate management.
Footnotes
Acknowledgement
We thank the Ninth Hospital of Nanchang for providing the various datasets.
Author contributions
Shian Yu wrote the manuscript and prepared the figures. Shian Yu and Hang Yuan analyzed the data. Shian Yu and Yong Cao analyzed the patient’s test results. Hang Yuan and Yong Cao critically revised the manuscript. Shian Yu and Yong Cao conceived and designed the study. All authors reviewed and approved the final manuscript.
Availability of data and materials
The data are not publicly available at the request of the patient, who wished for no personal data to be identifiable in any way.
Declaration of conflicting interests
The authors declare that there is no conflict of interest.
Ethics statement
All of the patient’s information has been de-identified. The reporting of this study conforms to the CARE guidelines. 14 Verbal consent for treatment and publication was obtained from the patient; therefore, we did not apply for ethics committee or institutional review board approval of the study protocol.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.