
Abstract
KAT6A syndrome is an autosomal dominant genetic disorder associated with intellectual disability due to mutations in the lysine acetyltransferase 6A (KAT6A) gene. There are some differences in phenotype between KAT6A gene variants. This current case report describes a 1-month-old male infant that had a nonsense mutation in the KAT6A gene. Neither of his parents had the mutation. The proband had feeding difficulties and a physical examination revealed the following: moderate dysphagia, hypoplastic laryngeal cartilage, poor audio-visual response, poor head-up ability, no active grasping awareness, microcephaly, high arched palate and he was significantly behind other children of the same age. Echocardiography showed that the foramen ovale was not closed. He was diagnosed with atrial septal defect (ASD) when 2 years old. The patient received ASD repair at 32 months of age. Head colour Doppler ultrasonography and brain magnetic resonance imaging showed cysts in the right ventricle and choroid plexus, which returned to normal at 2 years of age. This current case demonstrates that immediate surgery should be considered in newborns with KAT6A syndrome presenting with a heart malformation. A new KAT6A syndrome phenotype is described in this current case report, which requires early diagnosis and treatment.
Keywords
Introduction
The lysine acetyltransferase 6A gene (KAT6A; OMIM:616268) belongs to the MYST family of lysine acetyltransferases. 1 It is associated with several tumours and biological processes such as leukaemia, triple-negative breast cancer, leukaemia cell proliferation, tumorigenicity and chemoresistance via acetylation epigenetics.2–6 KAT6A protein may regulate gene transcription and a large number of protein modifications because of its modifiable lysine residues. 6 KAT6A protein plays an important role in development. 7 KAT6A gene knockout mice show embryonic lethality and homozygous deletion of KAT6A in mice results in high penetrance of ventricular septal defects. 8 To date, there have been nearly 100 cases of KAT6A gene variants reported; and their main phenotypes were developmental delay, intellectual disability, oromotor dyspraxia, characteristic facial features and language developmental delay.9–12 Different variants of KAT6A syndrome present with different phenotypes such as congenital heart disease, behaviour, immunity and sleep disturbances.1,9,11,13–16 This current case report describes a Chinese boy with a nonsense mutation in the KAT6A gene. In addition to the common phenotypes such as intellectual disability, oromotor dyspraxia and language developmental delay, he also had atrial septal defect (ASD). This current case report provides a reference for the diagnosis and treatment of KAT6A syndrome.
Case report
On 4th March 2019, the proband, a male newborn with a normal birth weight of 2.7 kg and a normal Apgar score, was born at 38 + 2 weeks of gestation. His mother reported no history of intrapartum hypoxia asphyxia or intrauterine distress. The mother had gestational diabetes mellitus and hyperlipidaemia during pregnancy. Two days after birth, the newborn was hospitalized in the Changsha Hospital for Maternal and Child Health Care, Hunan Normal University, Changsha, Hunan Province, China for 10 days due to ‘neonatal infection and congenital heart disease’ and then he was discharged home. At 3 months of age, the proband was hospitalized in the Changsha Hospital for Maternal and Child Health Care, Hunan Normal University for feeding problems. Whole exome sequencing found a KAT6A nonsense mutation (NM_006766.4: c.3070C > T: p.R1024X), but his parents did not have the mutation (Figure 1). The following diagnosis was considered: autosomal recessive 32 (OMIM: 616268)-related, developmental delay, moderate dysphagia, congenital laryngeal stridor and laryngeal cartilage dysplasia. At 5 months of age, the proband had poor laughing ability, poor audio-visual response, poor head erection, poor head-up ability, poor forearm support, no active grasping awareness and his development was significantly behind children of the same age. Considering the diagnosis of developmental delay, he was admitted to the Changsha Hospital for Maternal and Child Health Care, Hunan Normal University for rehabilitation training. His height at the time of training (aged 5+ months) was 60.0 cm, his head circumference was 38.7 cm and his body weight was 5.2 kg. He had a short stature, short limbs, a flat face, low nose bridge, wide eye distance, small cleft eyes, low ear position and a high arched palate (Figure 2). There was symmetrical breathing of both lungs without rales. Heart grade II/VI murmur could be heard in the anterior area. The abdomen was soft and the liver and spleen were not under the ribs. No obvious abnormality was found on auscultation. A specialist paediatric physical examination found the following: clear mind, average response, poor eye-tracking and listening, poor facial response, poor laughing and vocalization, little active pronunciation, crying and fussing, head upright < 5 s, head-up < 45°, poor elbow support, will not rollover. Poor lower body weight support. The muscle tone of the limbs was low and tendon reflexes can be elicited.

Whole genome sequencing identified a disease-causing gene mutation in the lysine acetyltransferase 6A (KAT6A) gene in the 3-month-old male infant proband but not in his family members: KAT6A NM_006766.4: c.3070C > T: p.R1024X. The colour version of this figure is available at: http://imr.sagepub.com.
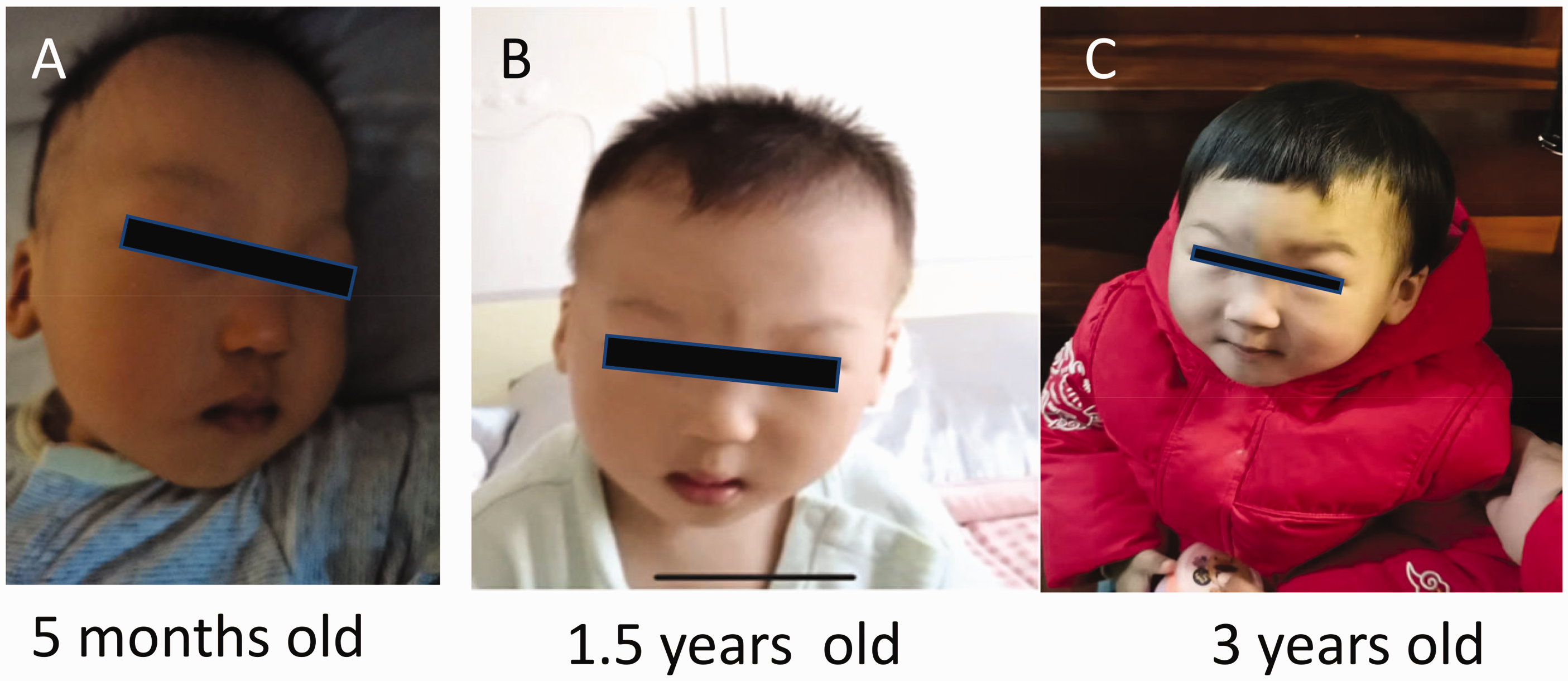
Representative photographs taken at different ages showing the characteristic facial phenotype in the male infant proband. The facial features included a flat face, low nose bridge, wide eye distance, small cleft eyes, low ear position and a high arched palate.
During the proband’s initial hospitalization in March 2019, a number of further examinations were undertaken. Cardiac ultrasound in March 2019 identified the following: ductus arteriosus left-to-right shunt (approximately 4.0 mm wide and 5 mm long), left-to-right shunt of the foramen ovale (approximately 4 mm wide) and mild tricuspid regurgitation (Figure 3). In March 2019, an approximately 4.9 mm wide dark area was found in the left renal pelvis by abdominal colour Doppler ultrasound. Brain colour Doppler ultrasound in April 2019 identified the following: intracranial flaky strong echo prosthetic valve endocarditis 1, multiple dark areas on the left choroid plexus and on two sides of the anterior horn of the bilateral lateral ventricles. Neonatal neurobehavioral test score in March 2019: 36 points.

Echocardiography undertaken before and after reparative surgery of an atrial septal defect (ASD) in a male infant. The patient received ASD repair under general anaesthesia with intubation at 32 months of age. (a) In March 2019, echocardiography showed the following: ASD (two-hole type), ductus arteriosus left-to-right shunt (approximately 4.0 mm wide and 5 mm long), left-to-right shunt of the foramen ovale (approximately 4 mm wide) and mild tricuspid regurgitation right atrium, right ventricle slightly larger, widened pulmonary artery, mitral, tricuspid and pulmonary valve regurgitation and (b) In February 2022, echocardiography post-ASD repair surgery showed a normal heart size with mild bi- and tricuspid regurgitation and cardiac changes.
After the proband was discharged from hospital in April 2019, he underwent a number of subsequent examinations. Brain colour Doppler ultrasound on 12 April 2019 identified the following: a cystic dark area adjacent to the anterior horn of the bilateral lateral ventricles (left 5.7 × 2.3 mm, right 8.4 × 3.8 mm, 6.4 ×3.3 mm); and there were multiple cystic dark areas in the left choroid plexus (Figure 4). Brain colour Doppler ultrasound on 24 June 2019 identified the following: bilateral lateral ventricle and third ventricle widened (anterior horn: left 4.6 mm, right 4.8 mm; body: left 4.7 mm, right 4.9 mm; third ventricle approximately 4.9 mm wide); and there were multiple cystic dark areas in the left choroid plexus (Figure 4). Cardiac ultrasound on 23 April 2019 identified the following: the foramen ovale was shunting from left to right (approximately 3.9 mm wide) and the heart rate was slightly faster. Cardiac ultrasound on 24 June 2019 identified the following from an atrial septal sound image: possible patent foramen ovale (echoic separation of approximately 4.0 mm could be seen at the fossa ovalis in the middle of the interatrial septum). Cardiac ultrasound on 28 April 2020 identified the following from atrial septal sonography: possible atrial septal defect and possible patent foramen ovale (echoes in the middle of the interatrial septum were interrupted by approximately 6.5 mm). Echocardiography on 26 November 2021 (Xiangya Hospital, Changsha, China) identified the following: congenital heart disease with atrial septal defect (two-hole type), slightly enlarged right atrium and right ventricle; widened pulmonary artery; bi-, tricuspid and pulmonary valve regurgitation. Colour Doppler echocardiography on 11 February 2022 (Xiangya Hospital, Changsha, China) identified the following: images consistent with the changes after ASD repair (Figure 3). Abdominal colour Doppler ultrasound on 24 June 2019 identified the following: separation of both kidneys and renal pelvis, 4.0 mm on the right and 2.9 mm on the left. Hip X-ray on 21 May 2020 identified the following: no obvious abnormality was found. Head magnetic resonance imaging (MRI) on 27 July 2019 (Hunan Children's Hospital, Changsha, China) identified the following: the right subependymal cyst was likely to be large, the bilateral lateral ventricles were slightly larger and the neck MRI showed no obvious abnormality. Cranial MRI on 25 May 2020 identified the following: no obvious abnormality was found in the routine sequence examination of head MRI. Electroencephalography (EEG) examination on 27 July 2019 (Hunan Children's Hospital, Changsha, China) identified the following: no epileptic waves were observed. A hearing test on 3 April 2019 recorded the following: 40 dnBl/50 dnBl. Repeated thyroid function, routine blood tests, liver and kidney function showed no obvious abnormalities. Haematuria genetic metabolism examination (i.e. hypothyroidism, phenylketonuria, congenital intellectual disability) on 27 July 2019 (Hunan Children's Hospital, Changsha, China) identified the following: no obvious abnormality was found. Chromosomal aneuploidy or genome copy number variants of ≥100 kb that are known to cause disease were not detected (Figure 5).

Colour B-ultrasound of the brain of a male infant showed brain abnormalities: (a) in April 2019, the images showed cystic dark areas adjacent to the anterior horn of the bilateral lateral ventricles and cystic dark areas of the left choroid plexus and (b) in June 2019, the images showed multiple cystic dark areas in the left choroid plexus and widened bilateral lateral ventricle and third ventricle.

Copy number variant sequencing results for the male infant proband showed that he did not have any copy number variants ≥100 kb that are known to cause disease. The colour version of this figure is available at: http://imr.sagepub.com.
The patient has been receiving the following at the Changsha Hospital for Maternal and Child Health Care, Hunan Normal University since May 2019: exercise training, EEG, magnetic therapy, biofeedback, language training, sensory integration training and mechanical exercise training. The patient received ASD repair under general anaesthesia with intubation at 32 months of age at Changsha Hospital for Maternal and Child Health Care, Hunan Normal University.
On 18 February 2022, at 3 years of age, his body weight was 10.9 kg, his height was 86.3 cm and his head circumference was 45.2 cm. The patient was climbing at 1 year and 6 months, standing alone at 2 years and 3 months and can now walk alone. He can go up and down two steps alone, but he is not stable. He can throw and kick a ball, walk backward, but he cannot run, cannot jump with both feet and he cannot walk a straight line within two lines that are 20 cm apart. At present, he has no meaningful pronunciation and occasionally pronounces the ‘ma’ sound unconsciously. He is mainly monophonic. He recognizes 4/6 real objects; he can build 4–5 layers of building blocks and he can doodle with a pen. Self-feeding with a spoon is messy and drinking milk from a cup easily results in choking. He can take socks off by himself, but he cannot use a zip, he cannot undress and he does not signal to defecate. The muscle tone of the limbs was slightly low and the tendon reflexes could be elicited.
The Ethics Committee of Changsha Maternal and Child Health Hospital approved this study (no. SOP-005-F02). Written informed consent was obtained from the patient's family for publication of the case details. The reporting of this study conforms to CARE guidelines. 17
Discussion
The KAT6 acetyltransferases play key roles in the regulation of transcription, various developmental processes, maintenance of haematopoietic and neural stem cells, regulation of haematopoietic cell differentiation, cell cycle progression and mitosis.7,18 Research has demonstrated that KAT6A protein plays a key role in cancer development and treatment, with inhibitors of KAT6A inducing senescence and arresting tumour growth. KAT6A is also involved in triple-negative breast cancer, ovarian cancer and haematological malignancies.2,6,19
Research has demonstrated that 51% of patients with KAT6A syndrome that have nonsense mutations also have cardiac malformations. 11 This is similar to the current case as he had ASD. In contrast to other cases, this current case presented with a new phenotype of a slightly enlarged right atrium and right ventricle, a widened pulmonary artery, and mitral, tricuspid and pulmonary regurgitation, which might have been caused by more than 2 years of ASD.10,13,20 The current findings suggest that if a KAT6A gene variant is found in a patient with a patent foramen ovale, then ASD repair surgery should be performed as soon as possible. An interesting phenomenon was observed during the monitoring of this current case. At the age of 1 month, brain B-ultrasound images showed cystic dark areas adjacent to the anterior horns of the bilateral lateral ventricles and multiple cystic dark areas on the left choroid plexus, suggesting that there were cysts in the brain ventricle and the left choroid plexus. Over time, the cysts disappeared and the bilateral ventricles and the third ventricle widened. The dark area of the lateral choroid plexus eventually disappeared and the brain structure returned to normal. These changes have not reported in other cases.
In the current case, there were typical features of KAT6A syndrome that included feeding difficulties, intellectual disability, microcephaly, hypotonia, language and developmental delay.9,11,12,14 However, there was no evidence of epilepsy, eye features, sleep disturbances, gastrointestinal problems, haematological and immunological disturbances.9,13 The most common clinical features of KAT6 syndrome are intellectual disability, speech delay and behavioural problems. 7 Currently, there is no effective treatment for KAT6 syndrome. Adeno-associated virus-mediated upregulation of the KAT6 gene could be a potentially effective therapeutic modality. The current patient has been undergoing language and motor training, but his most recent (aged 3 years and 4 months) Gesell developmental test report from August 2022 showed the following: 42 points for adaptation; 42 points for gross motor; 45 points for fine motor; 27 points for language; social 39 points. These test results indicate the equivalent of 17 months of development so he has moderate developmental delay. The rehabilitation training does not appear to have had a significant therapeutic effect on his developmental delay.
The p.R1024X mutation that was identified in this current case occurred in the first half of the acidic domain. As a consequence, the protein was terminated early and most of the acidic and Ser/Met domains were deleted, thus forming a dominant-negative mutation. Since KAT6A is an acetylated epigenetic modifier, different variants have different phenotypes in different genetic backgrounds.1,9,11,13–16 The mechanism of how the p.R1024X mutation leads to ASD and other phenotypes through an epigenetic mechanism needs to be studied further.
Footnotes
Acknowledgements
We thank the patient and his family for agreeing to participate in the present study and all those that helped us complete the research.
Declaration of conflicting interests
The authors declare that there are no conflicts of interest.
Funding
The authors disclose receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by grants from the Scientific Research Project of Hunan Education Department (no. 21A0043) and the Changsha Science and Technology Department (no. KQ1901012).