
Abstract
Primitive neuroectodermal tumor (PNET) of the lung is rare in adults, and treatment options vary. We herein describe the disease course and follow-up of PNET in an adult. A 27-year-old man was admitted to our hospital because of cough and headache, and whole-exome sequencing revealed positive expression of the EWSR1-FLI1 fusion gene and amplification of the APC gene. Although the patient received multidisciplinary treatment including chemotherapy regimens of etoposide plus cisplatin; focal radiotherapy focusing on the cerebrum, lung, and kidneys; and a subsequent palliative gastrointestinal operation, he eventually died of multiple organ functional failure. His overall survival period was 18 months, and his progression-free survival period was 4 months. During the treatment, the patient showed remarkable sensitivity to radiotherapy. In conclusion, PNET of the lung in adult patients is extremely rare, and the prognosis is very poor. Involvement of a multidisciplinary team in the development of personalized therapeutic strategies is essential. This patient with APC gene amplification showed excellent sensitivity to radiotherapy for intrapulmonary and intracranial lesions, suggesting that APC gene amplification may be related to radiotherapy sensitivity. However, further clinical research is needed.
Keywords
Introduction
Primitive neuroectodermal tumor (PNET) is an anaplastic malignant tumor consisting of undifferentiated small and round cells derived from the central nervous system and its surrounding musculoskeletal tissues. As a member of the Ewing sarcoma family, PNET can be classified into central or peripheral subtypes, both of which can affect organs other than those in the neurological, soft tissue, and skeletal systems in children and adolescents.1–4 The diagnosis of PNET largely depends on pathological and molecular examinations. Given the rarity and extremely poor prognosis of PNET with pulmonary invasiveness, 1 we believe that it is clinically valuable to report the clinical features, treatments, follow-up procedures, histological characteristics, and genomic variants of such cases to expand the knowledge of this disease. We herein contribute to previously reported cases of PNET by presenting a unique case of pulmonary, intracranial, and renal metastatic PNET with APC gene amplification with emphasis on the radiotherapy sensitivity of intracranial and intrapulmonary lesions. Our reporting of this case conforms to the CARE guidelines. 5
Case report
A 27-year-old man was admitted to our hospital with a 2-week history of cough, headache, and right hemiparesthesia. He had been previously healthy and had no family history or genetic disease history. He had received no previous medical treatments. The breath sounds in the patient’s left lung were reduced. Full-body imaging revealed an irregular, unevenly contrasted soft mass (15.17 × 9.16 cm) with a well-defined border in the left lung and nodules in the right lung. Other extrapulmonary lesions were also found, including a solid-cystic mass in the left occipital lobe, multiple masses in the bilateral kidneys, high density in the left adrenal gland, and low density in part of the thoracic vertebrae (Figure 1).

(a–c) A large and well-defined lobular soft tissue mass with uneven internal density contrast revealed by thoracic computed tomography. (d) An irregular solid-cystic mass in the left parietal lobe with uneven internal contrast and peripheral edema zone revealed by skull magnetic resonance imaging and (e, f) Multiple lesions in bilateral kidneys with uneven density.
Histopathological examination of a biopsy sample taken from the left lung mass by puncture under ultrasound guidance revealed diffuse distribution of small round intrapulmonary cells with hyperchromatic nuclei. Immunohistological staining of the samples was positive for CD56, CD99, vimentin, and Ki67 (approximately 30%) and negative for GFAP, CK, TTF-1, Syn, CgA, BCL-2, and EMA (Figure 2). These findings were consistent with the features of PNET. Whole-exome sequencing data from the lung lesion showed EWSR1-FLI1 gene fusion with enrichment of 63.79% and amplification of the APC gene (copy number, 3.24) in chromosome 5q22.2 (Figure 3). Collectively, these findings led to the diagnosis of metastatic PNET with an extremely poor prognosis accompanied by involvement of the brain, lung, bilateral kidneys, and abdominal cavity.
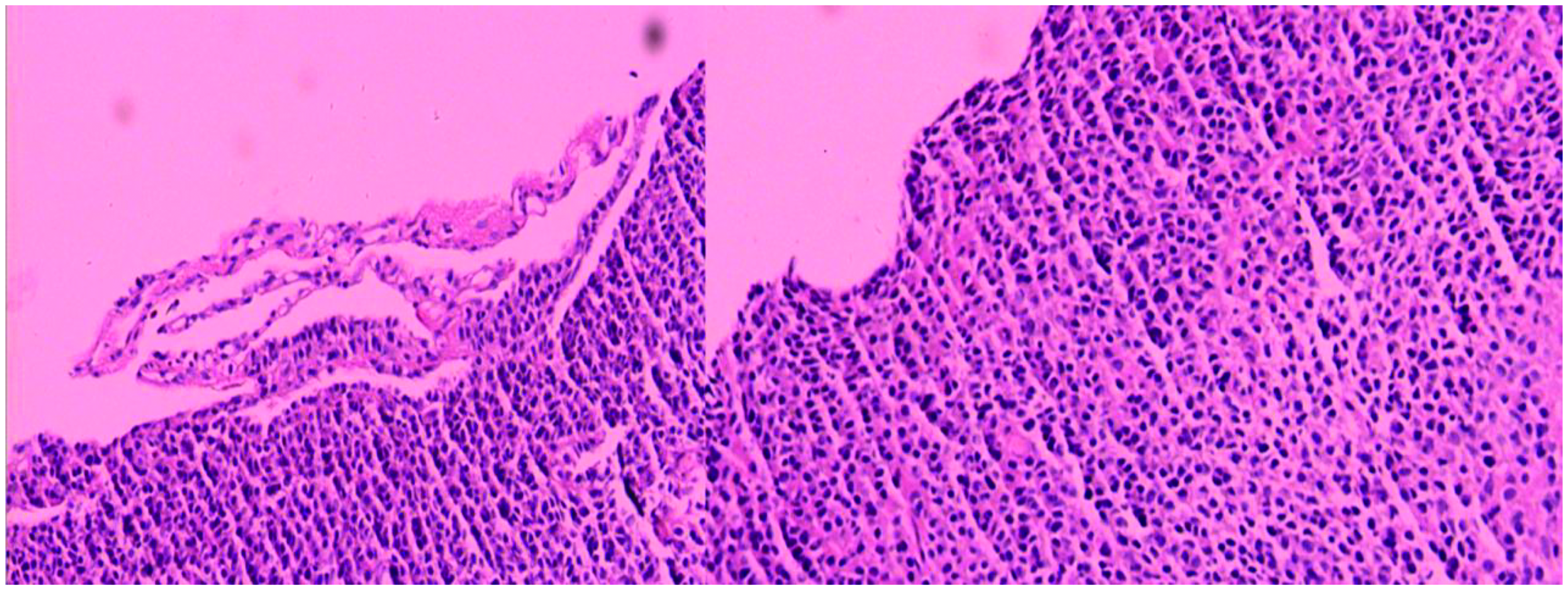
Numerous small, round/oval, and undifferentiated cells with hyperchromatic nuclei revealed by lung biopsy under microscope.
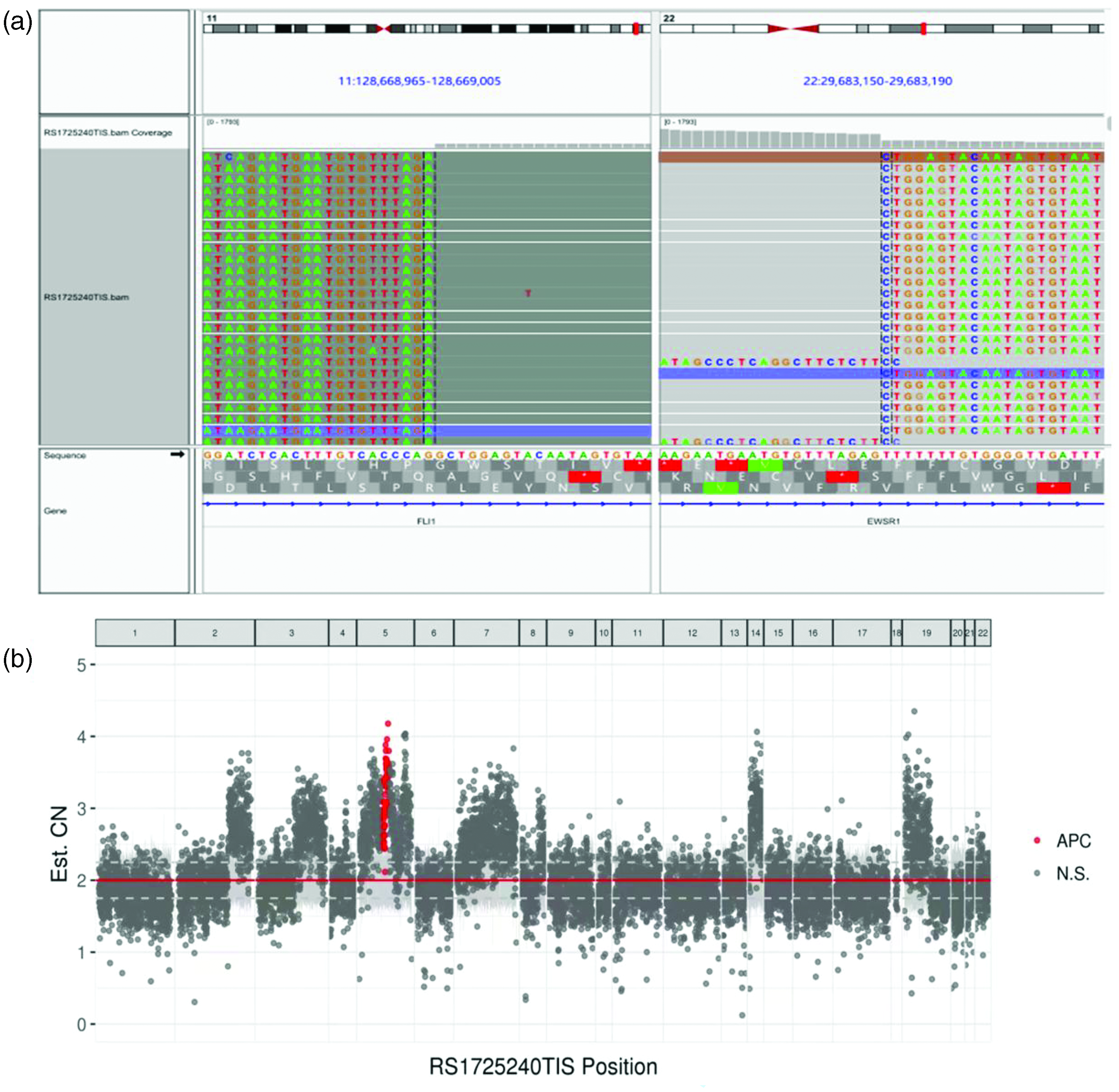
(a) Whole-exome sequencing indicated EWSR1-FLI1 (F5:E9) fusion gene with enrichment of 63.79% and (b) DNA copy number amplified for APC gene was 3.24 at chromosome 5q22.2.
First-line EP chemotherapy (etoposide at 85 mg/m2 on days 1–5, cisplatin at 75 mg/m2 on day 1) was administered. Intracranial radiotherapy of 3600 cGray for 12 cycles was also performed to alleviate symptoms of high intracranial pressure. The tumor size markedly decreased (Figure 4), and tremendous improvement in the patient’s headache, vomiting, and muscle weakness was achieved.

Significant reduction of tumor size and alleviation of peripheral edema after intracranial radiotherapy.
The patient’s tumor was identified as progressive during therapeutic response evaluation after three cycles of the EP chemotherapy regimen, at which time he was found to have advanced development of the lung lesion and progression-free survival (PFS) of 4 months. Focal radiotherapy in the left lung field was ordered (5000 cGray for 25 cycles), and the tumor substantially decreased in size (Figure 5). Unexpectedly, however, the patient reported abdominal pain and distension, vomiting, and constipation. Abdominal computed tomography revealed right-side colonic intussusception, a high-density lesion in the ileocolonic lumen, and a neoplastic mass at the bottom of the pelvis, which was attributed to metastasis. A palliative operation was performed to resolve the intestinal obstruction due to intussusception.

Significant reduction of lung lesions/tumors after intrapulmonary radiotherapy.
After the patient’s postoperative recovery, radiotherapy (1800 cGray for nine cycles) was administered to his left kidney field but failed to reduce the tumor size. The lesion in the lung thereafter continuously progressed with persistent fever and escalating pain in the left chest. The patient died of multiple organ dysfunction and failure with an overall survival (OS) period of 18 months.
Discussion
PNET is more commonly seen in children and adolescents than in adults. It is a high-grade malignant tumor with early metastasis and an extremely poor prognosis. PNET can reportedly develop anywhere in the body, including the orbit, nasal cavity, mediastinum, stomach, bone, uterus, kidney, neck, chest, pancreas, gingiva, paraspinal region, ovaries, breast, testes, and thymus;1–4,6–9 however, it is rarely found in the lung. Only 15 related cases were retrieved from the literature from 2012 to 2022 (Table 1). To contribute to the currently available real-world data, we have herein reported this special case of PNET with pulmonary, intracranial, and kidney lesions; APC gene amplification; and different radiotherapy sensitivities at different lesion sites.
Previous reports of patients with primary pulmonary primitive neuroectodermal tumor.

A&W, alive and well; CT, chemotherapy; DOD, died of disease; LL, left lung; LLL, left lower lobe; LUL, left upper lobe; RLL, right lower lobe; RML, right middle lobe; RUL, right upper lobe; XRT, radiation therapy; RL, right lung.
The clinical manifestations of PNET vary depending on the affected organ. When patients have lung-invasive tumors, they often exhibit nonspecific symptoms such as a productive cough, chest pain, and hemoptysis. In such patients, a large, highly invasive solid-cystic mass with uneven density and asymmetric contrast can be found by pulmonary imaging examination. Magnetic resonance imaging can reveal a low-signal image in muscular tissue. 10 The mass may be solitary or multiple, of various sizes, or lobulated. However, positron emission tomography–computed tomography reveals high uptake of 18F-fluorodeoxyglucose in the tumor and regional lymph nodes, where the standardized uptake value is significantly higher than that in peripheral healthy tissues. 11
The pathological features of PNET include highly aggressive behavior and small, round tumor cells originating from the central nervous system and sympathetic nervous system. 12 Differential diagnoses should include other small round cell tumors such as rhabdomyosarcoma, small cell lung cancer, lymphoma, and poorly differentiated synovial sarcoma. Frequently used specific immunohistological markers are CD9, vimentin, MIC2, FLI1, neuron-specific enolase, synaptophysin, and S-100.13,14 The expression levels of these markers are correlated with the stages of neuroectoderm differentiation.
The Ewing sarcoma family of tumors is genetically characterized by specific chromosomal translocations resulting in fusion of the EWSR1 (22q12) gene with one member of the ETS family of transcription factors,13–18 which can be determined by polymerase chain reaction and fluorescence in situ hybridization. APC gene mutation may also contribute to the pathogenesis of PNET through the WNT/wingless signal pathway. 19 Other studies have shown that mutation in the TP53, SK11, 15 PTCH, SMOH, 20 and MIC2 (CD99) 21 genes might be involved in the pathogenesis of PNET.
The reported routine treatment strategies for PNET include combinations of surgery, radiochemotherapy, and novel exploratory target therapy. Standard chemotherapy regimens consist of etoposide, vincristine, doxorubicin, and cyclophosphamide. Chen et al. 22 examined 31 patients with Ewing’s sarcoma of the skull and/or peripheral PNET and found a median PFS of 8 months and OS of 22 months; the OS rates at the 1-, 2-, and 5-year time points were 74.2%, 34.9%, and 13.1%, respectively. Additionally, surgery and radiotherapy can significantly improve the prognosis, especially for patients with intracranial and skeletal involvement.6,23 For those with renal PNET, postoperative adjuvant chemotherapy is more beneficial than single surgical treatment as measured by OS. 24 Therapies targeting gene mutations, such as anlotinib and pazopanib, can moderately improve the therapeutic response.16,25 However, a more standardized and precise treatment strategy is desired, and more evidence-based medical developments are required to achieve this goal.
In the present case, a patient with expression of the EWSR1-FLI1 fusion gene and APC gene amplification received three cycles of the EP chemotherapy regimen and head-, thorax-, and kidney-field radiotherapy with alleviation of headache, vomiting, and muscle weakness followed by palliative abdominal surgery. He ultimately reached PFS of 4 months and OS of 18 months. Notably, his intracranial and intrapulmonary lesions were sensitive to radiotherapy, but his abdominal and kidney lesions were not; this may be attributed to the heterogeneity of PNET with multiple organ infiltration. The patient’s clinical progression suggests a possible relationship between APC gene amplification and radiotherapy sensitivity, which requires further clinical research.
Conclusion
PNETs in adults have been rarely reported in the literature and are extremely rare in patients with gene mutation characteristics and complete clinical follow-up. Thus, more cases of PNETs and long-term follow-up studies are warranted to fully understand PNETs in the adult population. The present case report might serve as an additional reference among the few available as guidance for clinicians and radiologists.
Supplemental Material
sj-pdf-1-imr-10.1177_03000605221118704 - Supplemental material for A rare radiotherapy-sensitive primitive neuroectodermal tumor with APC gene amplification in an adult: a case report and literature review
Supplemental material, sj-pdf-1-imr-10.1177_03000605221118704 for A rare radiotherapy-sensitive primitive neuroectodermal tumor with APC gene amplification in an adult: a case report and literature review by Xin He, Shan Song, Peidan Yang, Feng Cao, Weijing Li and Ping Liang in Journal of International Medical Research
Footnotes
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Ethics statement
Written informed consent for publication of this case report and the accompanying images was obtained from the patient. The study was approved by the Fourth Hospital of Hebei Medical University Ethics Committee (approval number: 2021KY071).
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.