
Abstract
In resource-constrained settings, mucopolysaccharidosis (MPS) is a rare hereditary metabolic illness that frequently remains undiagnosed. We present a scenario that illustrates the challenges in diagnosing and managing MPS because of test inaccessibility, and we propose potential approaches to minimize the hurdles. We recommend that physicians anticipate a rare genetic disease, such as MPS, based on the clinical history findings from routine radiological investigations. Additionally, stakeholders should perform risk stratification and implement screening tests as soon as possible to ensure that patients are effectively enrolled in treatment programs.
Keywords
Introduction
Mucopolysaccharidoses (MPS) are a group of rare genetic disorders characterized by a deficiency or dysfunction of enzymes that metabolize glycosaminoglycans (GAGs). They account for less than 0.1 percent of all genetic disorders.1,2 Different MPS types exhibit comparable symptoms, especially MPS I and II; however, there are some differences, such as severe neurological issues in MPS III and hydrops fetalis in MPS VII. 3 These clinical signs facilitate diagnosis, but early and precise diagnosis in the asymptomatic stage is critical for better outcomes.3,4 On the basis of experimental and clinical investigations, hematopoietic stem cell transplantation (HSCT) and enzyme replacement therapy (ERT) have been established in recent decades. 5
MPS is divided into 7 types and 11 subtypes differentiated biochemically by the related enzyme deficiency. MPS II or Hunter syndrome is the only X-linked-inherited mucopolysaccharidosis primarily affecting men, with an incidence rate ranging from 0.38 to 1.09 per 100,000 live male births. However, no documented study in Bangladesh shows the prevalence of Hunter’s disease. 6 Moreover, both MPS I and II present with hepatosplenomegaly, cognitive retardation, dysostosis multiplex, dysmorphic appearance, corneal clouding, and cardiac abnormalities. Additionally, MPS II shows dermatological signs. 7 MPS III has limited central nervous system involvement features and often presents with dysostosis multiplex and dysmorphic appearances.8,9
Confirmatory diagnosis of MPS can be made by urinary and blood GAG tests, enzyme assays, and gene tests. Moreover, it can be diagnosed antenatally by fibroblast enzymatic activity in amniotic fluid and marked GAG storage in fetal organs.10,11 Physicians are confronted with difficulties in identifying patients with MPS in the early stages of the disease before irreversible damage has occurred. 12 Moreover, most enzymatic studies are unavailable in resource-limited centers, such as Bangladesh. Hence, a careful and critical approach with supportive imaging, such as a skeletal survey, following the patient’s clinical features is essential for diagnosing MPS types.13,14 Attempts have been made to diagnose MPS types without biochemical tests, including a mnemonic screening tool for MPS II developed by the Hunter Outcome Survey group. 15 Additionally, researchers from Nepal diagnosed MPS II from history, clinical features, and skeletal survey as they lack biochemical testing facilities in their limited-resource settings.13,16
Managing MPS involves preventing severe disease and enhancing the quality of life. Patients have several therapy options, including recombinant human alpha-L-iduronidase in patients with MPS I and intravenous ERT with recombinant human iduronate-2-sulphatase-idursulfase, HSCT, anti-inflammatory treatment, and palliative care with symptomatic surgeries for MPS II.3,17,18 Although there is no approved treatment for MPS III, ERT with Mepsevii (vestronidase alfa-jvbk) can be used to treat MPS VII.5,19 Many of these therapies, especially ERT, show potential to significantly improve the outcome and quality of life if started early in the disease course. 5
In Bangladesh, the therapeutic options for MPS are limited. Most cases are treated with supportive management, such as nonsteroidal anti-inflammatory drugs and vitamin D supplements. Furthermore, because of the lack of enzymatic assays, ERT is not possible. Over the previous decade, Bangladesh has developed a few HSCT centers, although no MPS-related transplants have been attempted.20,21 In agreement with the CARE reporting checklist, we present the following case to highlight the challenges of addressing a rare metabolic illness in Bangladesh, enabling clinicians and stakeholders to respond appropriately. 22
Case presentation
An 11-year-old South Asian boy presented to the pediatric in-patient department with lethargy, poor feeding for 2 months, and respiratory distress for 2 days. The patient had a history of difficulty swallowing both liquid and solid foods that developed over 2 months, leading to severe dehydration. According to the parent’s statement, they noticed a delay in his developmental milestones for the first time when he could not sit properly, even at the age of 9 months. Afterward, a history of repeated respiratory infections was mentioned, although he never required hospital admission. The parents also complained of the child’s disturbed sleep pattern and agitated behavior. He was unable to communicate verbally and never attended any school. However, the patient’s attendants mentioned that before this episode of hospitalization, he used to have a good appetite and could sit and walk some distance independently.
He had not developed any bowel or bladder control to date. There was no significant family history, and none of his siblings were affected. The patient had no history of convulsion, loss of consciousness, bleeding manifestation, jaundice, vomiting, constipation, or diarrhea.
There was a manifestation of growth retardation and coarse facial features (Figure 1a). The patient had an occipitofrontal circumference of 58 cm, frontal bossing, thick eyebrows, depressed nasal bridge, broad lip, malocclusion of teeth (Figure 1a), oral ulcer, short neck, and tiny stubby fingers (Figure 1c, d). No clouding of the cornea was observed. An expert opinion from the ophthalmology department was obtained, and no abnormality was detected in the cornea, fundus, or lens. The patient had hepatomegaly, with the liver measuring 6 cm from the subcostal margin in the right midclavicular line, a palpable spleen, and an umbilical hernia (Figure 1b). Palpatory and auscultatory findings of bronchopneumonia were also present.

Clinical presentation of the patient reported in this case. (a) Facial features, (b) umbilical hernia, and (c, d) small stubby fingers of hand and feet.
Regarding imaging features, a chest X-ray revealed spatulated ribs (tapering of the posterior end of the rib). The right dome of the diaphragm was 4 cm higher than the left (Figure 2a). A hand wrist radiograph showed proximal tapering of metacarpal bones (bullet shape) and fusion of interphalangeal joints (Figure 2b). The clinical aspects of this case are summarized in Table 1.
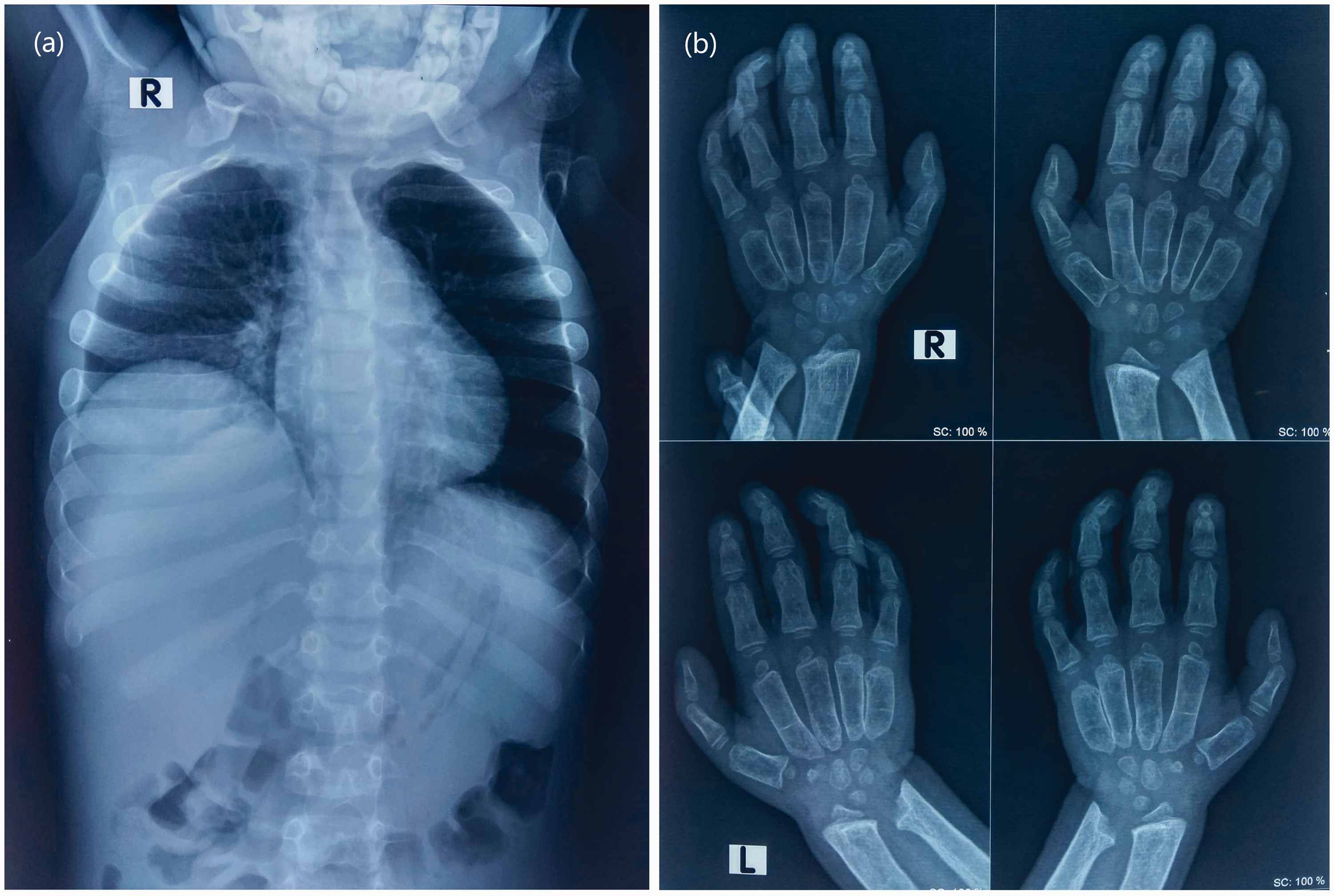
Imaging findings. (a) Chest X-ray posteroanterior view showing spatulated ribs and (b) X-ray of hand and wrist joint showing proximal tapering of metacarpal bones (bullet shape) and fusion of the interphalangeal joint. Upper panels: right hand and wrist joint. Lower panels: left side.
Presenting features of the case compared with the features of MPS I, II, III, and VII.

“+” = Present, “–” = Absent.
MPS, mucopolysaccharidosis; OFC, occipitofrontal circumference.
Structure of this table is modified from the work of Burton, B.K., Giugliani, R. Diagnosing Hunter syndrome in pediatric practice: practical considerations and common pitfalls. Eur J Pediatr 171, 631–639 (2012). 23
After receiving written informed consent from the patient’s guardian, the patient was scheduled for regular investigations, which revealed severe hypernatremia (168 mEq/L) with hyperchloremia (132 mEq/L). Complete blood count, thyroid-stimulating hormone, and free T4 were within normal ranges. Ultrasonography of the whole abdomen showed no abnormality, and kidney and liver function were normal. Because of the financial constraints and lack of testing facility, no urinalysis and echocardiography were performed. The clinical assessment and radiological findings revealed a wide variety of diagnostic possibilities for several forms of MPS, primarily I, II, III, and VII. All information and images were collected with prior written informed consent from the patient’s guardian.
Discussion
This case report describes the case of an 11-year-old boy diagnosed with probable MPS II from the clinical correlations and radiological findings in a resource-limited setting where the confirmatory biochemical tests were not available. Infants with attenuated MPS appear relatively normal at birth, and early developmental milestones may be typical. Furthermore, some signs and symptoms overlap with common childhood complaints, and MPS often remains undiagnosed until later in childhood. 23 In our case, the patient showed a delay in reaching his early developmental milestones and developed characteristic coarse facial features with repeated visits to the primary care center for recurrent respiratory tract infection. The possibility of a rare and genetic disease could not be investigated, even after the presence of multiple signs 23 that a single illness could explain his presentation.
The presentation of this case is consistent with the characteristics of MPS I and II shown in Table 1. However, he did not have any corneal change consistent with his MPS II diagnosis. 23 Previous cases with tonsillitis, adenoids, polyp presence on the laryngeal inlet, polypoid lesions, supraglottic narrowing, thickening of the tongue, and oropharyngeal mucosa due to the deposition of GAGs explain the dysphagia and sleep apnea in our patient. 24 Structural involvement of his heart and brain could not be detected because no echocardiography, computed tomography, and magnetic resonance imaging tests were performed. A screening test to diagnose MPS biochemically (urine for GAG test) and an enzyme assay were initially planned. However, they could not be implemented because of the unavailability and financial constraint of the family. After 9 days of his hospital stay, the family was discharged against medical advice.
Although this patient did not have the financial capability to afford definite treatment of the disease and thus only received supportive care, the rationality of treating this patient with ERT or HSCT remained open to debate because the efficacy of ERT and HSCT is presumably better in the early stages of the disease. 25 However, previous studies noted that patients with MPS II, irrespective of the age of presentation and central nervous system involvement, should receive ERT to potentially reduce disease severity. 26
This case depicts a dilemma for the patient, his family members, and the physicians. The lack of a testing facility led to delayed diagnosis at an older age, which limits the effectiveness of available treatments because the irreversible changes have already occurred. Therefore, improved disease recognition and early diagnosis through precise clinical evaluation, especially in resource-limited settings, are essential. The stakeholders at resource-limited stages should follow the guidelines to categorize patients with possible MPS into high-risk and low-risk groups to ensure that screening tests (urine for GAG test) at an early age can be implemented. 12 In turn, this will help patients benefit from novel therapeutics, HSCT, or ERT, which are likely to increase survival rates while significantly improving their quality of life. Moreover, resource-rich countries should invest more in research to make testing facilities cost-effective and thus available in countries with limited resources. More treatment options that are sustainable and effective, especially in late-diagnosed individuals, should be made available through novel research and pragmatic implementation.
Supplemental Material
sj-pdf-1-imr-10.1177_03000605221106412 - Supplemental material for Limited diagnostic facilities impeding the therapeutic approach of Mucopolysaccharidosis in Bangladesh: a case report
Supplemental material, sj-pdf-1-imr-10.1177_03000605221106412 for Limited diagnostic facilities impeding the therapeutic approach of Mucopolysaccharidosis in Bangladesh: a case report by Orindom Shing Pulock, Susmita Dey Pinky and Syeda Humaida Hasan in Journal of International Medical Research
Footnotes
Acknowledgements
We thank our patient’s parents for their cooperation and for providing consent for publication, and we appreciate the continuous support from the Department of Pediatrics, Chattogram Medical College Hospital.
Author contributions
All authors contributed to the study conception and design. Material preparation, data collection, and manuscript writing were performed by OSP and SDP, and they contributed equally to this article. SHH coordinated the project.
Ethics statement
Ethical approval for case reports and case series is waived by our institutional ethical review committee. This research also adheres to the standards of the Helsinki Declaration. Written informed consent was obtained from the parent of the patient to publish this report in accordance with the journal’s patient consent policy.
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.