
Abstract
The purpose of this review is to create more awareness regarding the epileptic manifestations of non-ketotic hyperglycaemia, which are not widely recognised, and to assist understanding of the pathophysiology involved. Given that type II diabetes is one of the common causes of morbidity worldwide, it is important to appreciate the various neurological manifestations of non-ketotic hyperglycaemia.
Here, I present two cases and review the existing literature. Both patients developed irreversible vision loss, which is a novel finding because only transient visual defects have previously been reported. The review includes a detailed discussion of the pathophysiology and characteristic magnetic resonance imaging (MRI) findings of patients with defects in cerebral lobar regions, which were associated with a variety of clinical manifestations. These manifestations can be ascribed to epileptic phenomena involving various parts of the cerebrum.
Hyperglycaemia can lead to the irreversible loss of vision. Early diagnosis and treatment on the basis of the clinical features and characteristic MRI findings are important to avoid an epilepsia partialis continua-like state and irreversible visual impairment.
Keywords
Introduction
Hyperglycaemia can cause encephalopathy, hemiparesis, hemisensory loss, focal seizures, and movement disorders, such as chorea, athetosis, and hemiballismus.1–4 In this review, I shall discuss the lesser-known symptoms of non-ketotic hyperosmolar hyperglycaemia, which are referable to various cerebral regions, and have a variety of clinical presentations. In addition, I shall describe two cases of irreversible cortical vision loss owing to non-ketotic hyperglycaemia (NKH), a phenomenon that has not been described previously.
The reporting of these cases conforms to the CARE guidelines. 5
Case 1
Patient information and clinical findings
A woman in her 60s was hospitalised because of transient visual symptoms in the form of flashes of colour lasting for few seconds several times a day for more than 2 weeks, followed by severe bilateral symmetrical loss of vision. Physical and neurological examinations revealed bilateral vision loss with no light perception. Her pupils were equal in size and reacted to light, and her optic discs were both normal.
Diagnostic assessment
The patient’s fasting and postprandial blood glucose concentrations at the time of admission were 13.6 mmol/L and 22.2 mmol/L, respectively, but serum ketones were absent. Her serum creatinine, urea, sodium, and potassium concentrations were 0.380 mmol/L, 40.5 mmol/L, 138 mmol/L, and 3.40 mmol/L, respectively. Her creatinine and urea concentrations were reduced to 0.230 mmol/L and 20.1 mmol/L by three cycles of haemodialysis. Her cerebrospinal fluid (CSF) was normal, but electroencephalography (EEG) showed a loss of the posterior occipital alpha rhythm.
Therapeutic intervention
Consent for treatment was obtained from the patient and her hyperglycaemia was immediately corrected by means of an insulin infusion.
Follow-up and outcome
There had been no improvement in the patient’s vision 6 months after the initial examination (Figure 1). This case has been described previously.6

Case 1 (first published as a Case of the Week in Am J Neuroradiol in March 2018) imaging results. (a) T2 sequence. (b) Fluid-attenuation inversion recovery shows bilateral occipital subcortical and diffuse pons hypointensity. (c) Diffusion-weighted image shows subtle restriction in the pons and bilaterally in the occipital cortex. (d) An apparent diffusion coefficient (ADC) sequence on magnetic resonance imaging shows corresponding areas of hypointensity. (e) Contrast T1 imaging shows bilateral occipital enhancement. (f) Normal angiographic findings.
Case 2
Patient information and clinical findings
A man in his 60s presented with frequent episodes of flickering bright colour in his right visual field that lasted for a few seconds to a few minutes and continued for more than 1 month, after which he experienced sudden-onset persistent loss of vision in his right hemi-field. He had also experienced a few partial motor seizures, involving transient gaze preference towards the right side and occasional episodes of right-sided facial spasm. Neurological examination revealed right homonymous hemianopia, but no other focal deficits.
Diagnostic assessment
The patient’s fasting and postprandial blood glucose concentrations were 14.4 mmol/L and 22.9 mmol/L, respectively, and his glycosylated haemoglobin (HbA1c) was 13%, but serum ketones were absent. His other biochemical and haematological parameters were normal. Interictal EEG showed intermittent slowing of the theta range in the left occipital region.
Therapeutic intervention
Consent for treatment was obtained from the patient, and his blood glucose concentration was brought under control by means of an insulin infusion and subsequent subcutaneous insulin injection. Perimetry revealed right homonymous hemianopia.
Follow-up and outcome
No improvement in the patient’s vision occurred, and findings consistent with this were made by repeat perimetry (Figures 4A and B) and brain magnetic resonance imaging (MRI) 2 years after discharge (Figures 2 and 3).
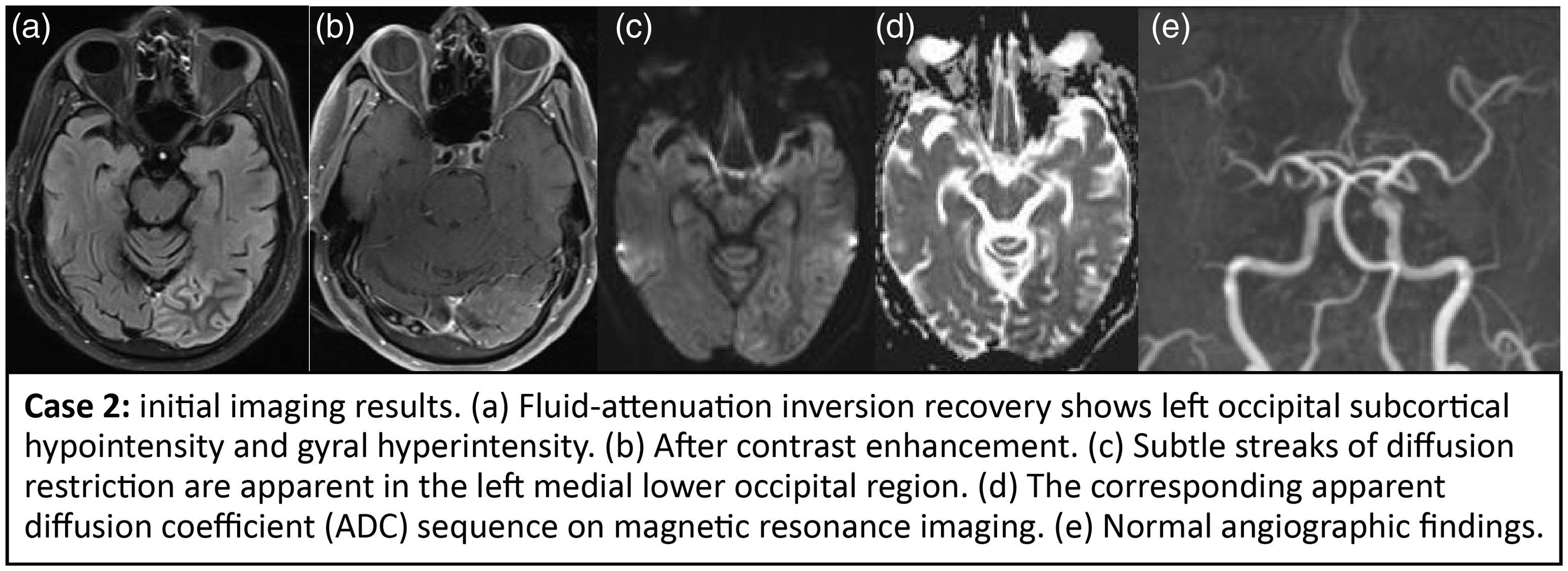
Case 2: initial imaging results. (a) Fluid-attenuation inversion recovery shows left occipital subcortical hypointensity and gyral hyperintensity. (b) After contrast enhancement. (c) Subtle streaks of diffusion restriction are apparent in the left medial lower occipital region. (d) The corresponding apparent diffusion coefficient (ADC) sequence on magnetic resonance imaging. (e) Normal angiographic findings.

Case 2: magnetic resonance imaging findings 2 years after the initial presentation. (a) Fluid-attenuation inversion recovery. (b) T2-weighted image, showing encephalomalacic changes, gliosis in the left occipital lobe, and ex vacuo dilatation of left lateral ventricle. (c) Diffusion-weighted image, showing no diffusion restriction. (d) Magnetic resonance angiography time-of-flight image, showing no significant stenosis of the intracranial vessels.

Case 2: perimetry findings. (A) Perimetry images obtained during the initial hospitalisation, showing right homonymous hemianopia. (B) Perimetry images obtained 2 years after the initial hospitalisation, showing persistence of the right homonymous hemianopia, but some improvement.
Epileptic presentations of non-ketotic hyperglycaemia
Transient visual symptoms (Tables 1, 2, and 3)
In NKH that predominantly affects the occipital lobes, visual complaints of various types are the most common manifestations. Flickering light, transient flashes of colour (occipital seizures), nystagmoid eye movements, transient conjugate eye deviation, transient bilateral visual loss, transient hemianopia, field defects,7–17 unformed or complex visual hallucinations,16,18–23 oscillopsia, metamorphopsia, and pallinopsia24–27 have been recorded.
Vision loss and or occipital seizures documented.

CT, computed tomography; fMRI, (functional) magnetic resonance imaging; SPECT, single-photon emission computerised tomography; EEG, electroencephalography; FLAIR, fluid-attenuation inversion recovery; HbA1c, glycosylated haemoglobin; BOLD, blood oxygen level-dependent imaging, DWI, diffusion-weighted imaging.
Complex hallucinations and delusions documented.

NAA, n-acetylaspartate; MRI, magnetic resonance imaging; MRS, magnetic resonance spectroscopy; CT, computed tomography; CSF, cerebrospinal fluid; HMPAO, D,L-hexamethylene-propyleneamine oxime; SPECT, single-photon emission computerised tomography; EEG, electroencephalography; FLAIR, fluid-attenuation inversion recovery; HbA1c, glycosylated haemoglobin; CVHs, complex visual hallucinations.
Unusual visual phenomena documented.

SWI, susceptibility-weighted imaging; DWI, diffusion-weighted imaging; CT, computed tomography; MRS, magnetic resonance spectroscopy; FLAIR, fluid-attenuation inversion recovery.
Irreversible visual loss (Table 4)
Irreversible vision loss documented.

HbA1c, glycosylated haemoglobin; MRI, magnetic resonance imaging; FLAIR, fluid-attenuation inversion recovery, DWI, diffusion-weighted imaging.
Non-visual symptoms (Tables 5 to 10)
Focal motor seizures (automatism; clonic or tonic types) and non-motor focal seizures can occur, the latter causing behavioural arrest or abnormalities such as psychosis or delirium.28–30 Cognitive seizures, such as aphasia, aphasic status epilepticus, and alexia; somatosensory seizures;16,31–37 and epilepsia partialis continua (EPC)38–40 can also be observed. Thus, focal seizures that involve normal awareness or impaired awareness, and reflex seizures41–50 can be caused by NKH.
Aphasic status epilepticus documented.
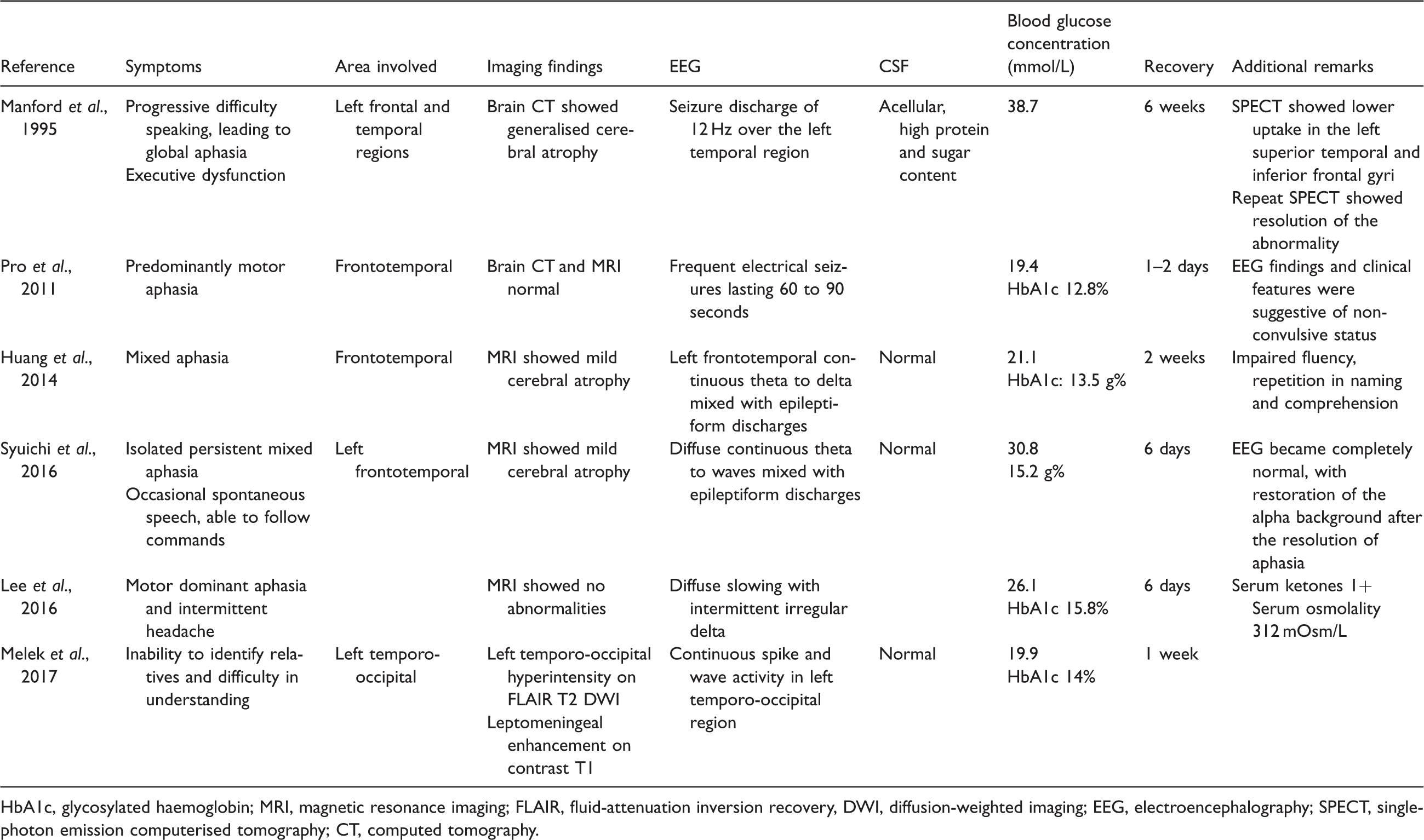
HbA1c, glycosylated haemoglobin; MRI, magnetic resonance imaging; FLAIR, fluid-attenuation inversion recovery, DWI, diffusion-weighted imaging; EEG, electroencephalography; SPECT, single-photon emission computerised tomography; CT, computed tomography.
Non-convulsive status epilepticus/frontal lobe dysfunction documented.

EEG, electroencephalography; NCSE, non-convulsive status epilepticus.
Non-convulsive status epilepticus/psychosis delirium documented.

HbA1c, glycosylated haemoglobin.
Non-convulsive status epilepticus/alexia without agraphia documented.

EEG, electroencephalography; FLAIR, fluid-attenuation inversion recovery; MRI, magnetic resonance imaging.
Epilepsia partialis continua documented.
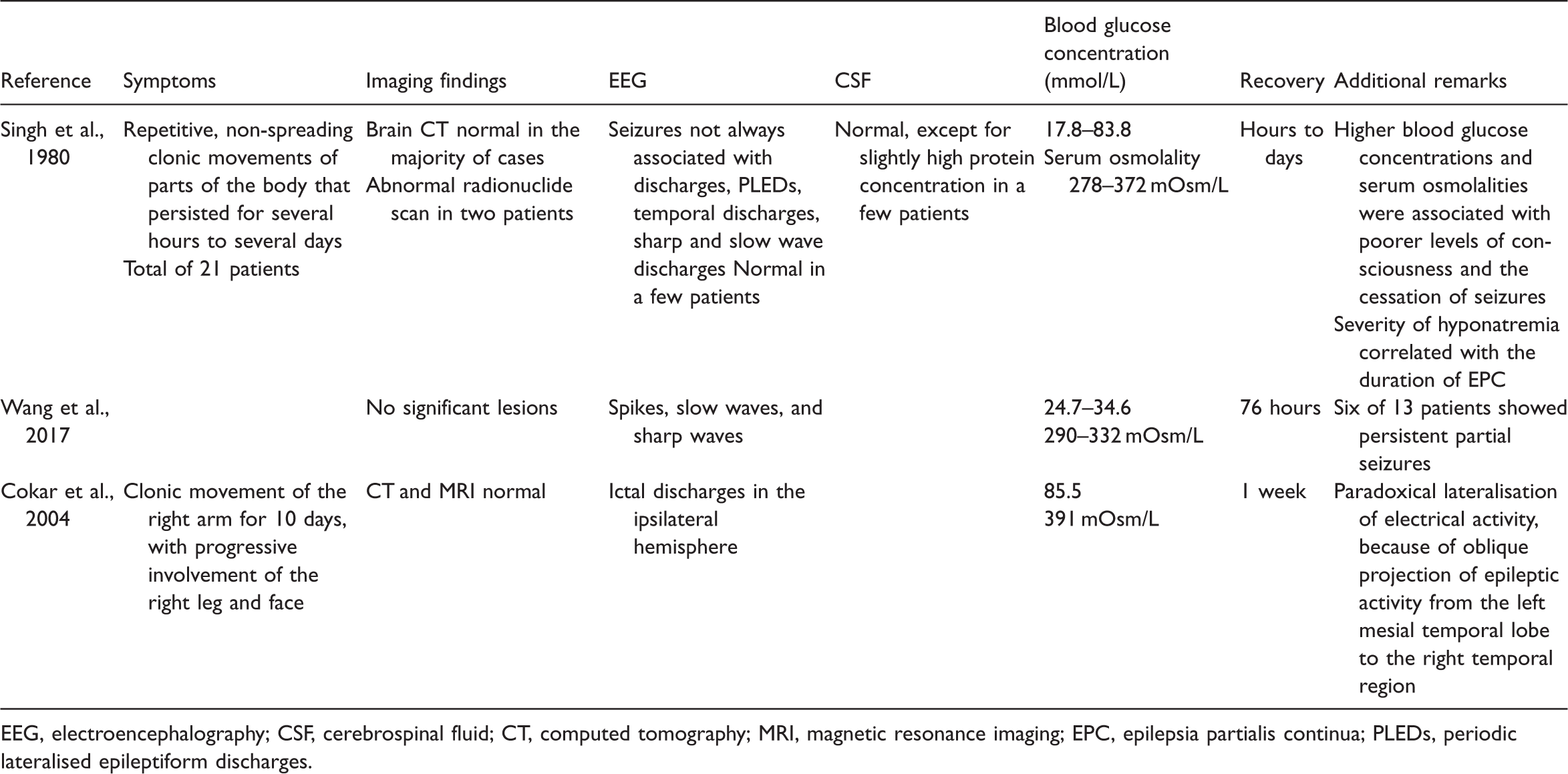
EEG, electroencephalography; CSF, cerebrospinal fluid; CT, computed tomography; MRI, magnetic resonance imaging; EPC, epilepsia partialis continua; PLEDs, periodic lateralised epileptiform discharges.
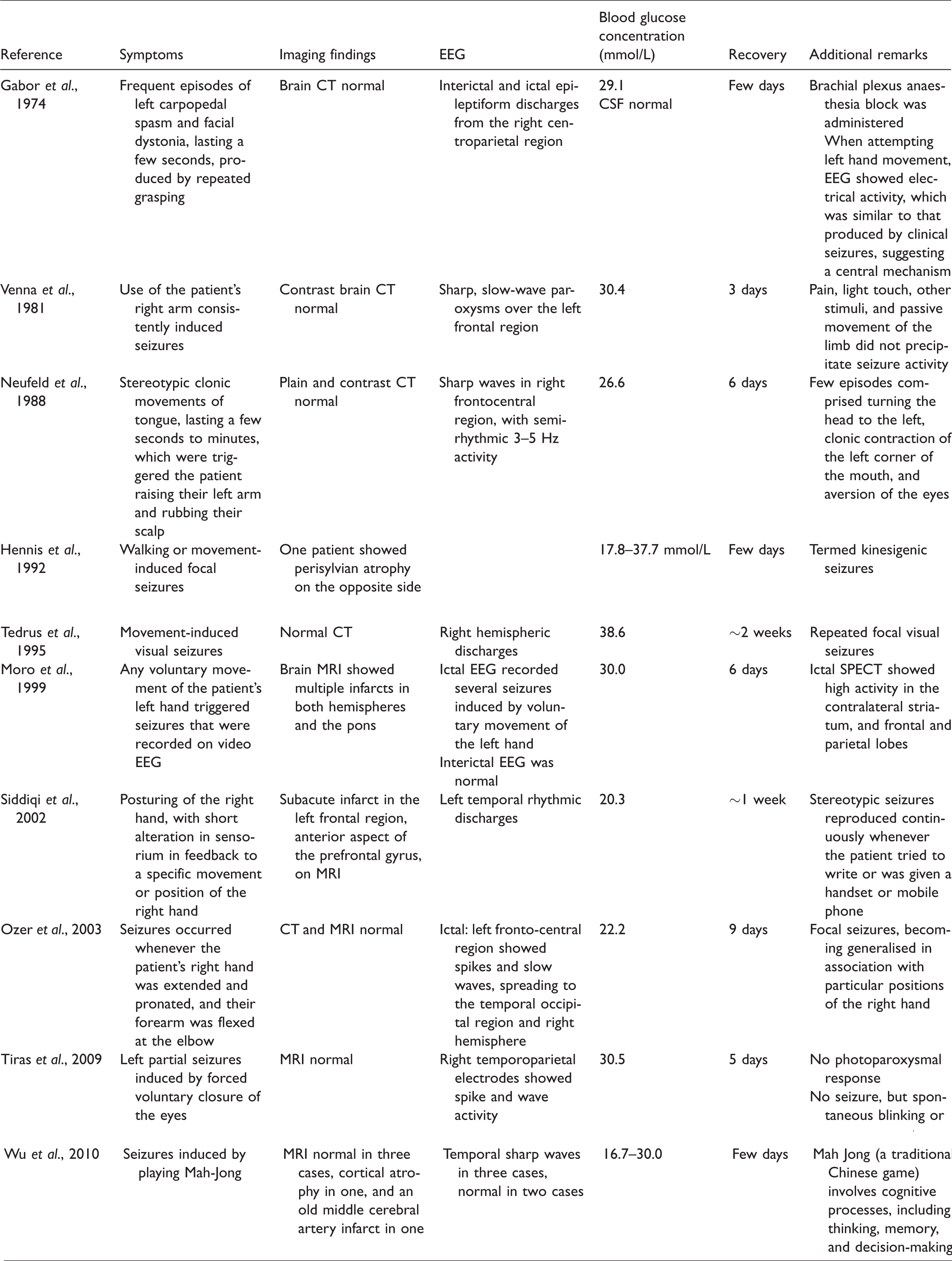
Reflex epilepsy documented.
Pathogenesis
A number of pathogenetic mechanisms have been postulated for the symptoms described above. Low concentrations of the inhibitory neurotransmitter γ-aminobutyric acid (GABA) can cause seizures, and in NKH, low Krebs cycle activity and brain glucose utilisation leads to greater metabolism of GABA to succinic acid to yield energy (the GABA shunt), causing GABA deficiency. In contrast, in ketotic hyperglycaemia, GABA concentrations are maintained by the activity of glutamic acid decarboxylase. 51 In addition, the utility of a ketogenic diet for seizure control may indicate that ketones help prevent seizures. 3
ATP-sensitive potassium channels are also thought to be responsible for neuronal hyperexcitability and the precipitation of seizures in hyperglycemia. 52 Astrocytes cultured in a high-glucose environment show low mRNA expression of the Kir4.1 potassium channel, but a restoration of the normal glucose concentration normalises the expression after a few days. In addition, glial glutamate uptake is low in a high-glucose environment. Thus, low glutamate uptake in combination with poor potassium clearance, because of low Kir4.1 expression, might lead to excitotoxic damage to neurons. 53
Previous studies have also suggested that disruption of the blood-brain barrier plays a role in the pathogenesis of the neurological signs described above. Gyral and leptomeningeal contrast enhancement has been observed, and may result from a disruption of the blood–brain barrier and extravasation of contrast medium, because of the greater metabolic activity during seizures. A delay in the gadolinium enhancement of the CSF space overlying the cortical region has also been observed using fluid-attenuation inversion recovery (FLAIR), and is suggestive of blood–brain barrier disruption. 54 This could be related to a delay in gadolinium enhancement on FLAIR imaging, which also occurs in patients who experience post-thrombolysis stroke. In this situation, early blood–brain barrier disruption occurs, which is also referred to as hyperintense acute reperfusion marker, and this is associated with haemorrhagic transformation and a poor clinical outcome. Iwata et al. demonstrated contrast enhancement of the globus pallidus prior to the development of homogenous hyperintensity on T1-weighted imaging, which is suggestive of blood–brain barrier destruction.55,56
The hypoxic ischemic state that is caused by hyperglycaemia could lead to excitotoxic axonal damage and the accumulation of free radicals or iron, causing a hypointense signal on T2-weighted images, 16 which could also be caused by intracellular osmotic dehydration.
The presence of diffusion restriction in many cases of NKH suggests that cytotoxic oedema may be an underlying pathogenetic mechanism. Classic posterior reversible encephalopathy syndrome (PRES), which also involves the parietooccipital region, has vasogenic oedema as an underlying mechanism, but this is associated with a hyperintense signal on T2-weighted images and diffusion restriction is not typical. 57
Central pontine myelinosis (CPM) has been documented in a patient during the treatment of hyperosmolar hyperglycaemia, which implies that osmotic demyelination may be involved in the pathogenesis. Oedema could lead to the compression of fibre tracts, leading to demyelination. The pons may become involved because of its tightly packed grey and white fibres, which are susceptible to osmotic demyelination. 58 However, the basal ganglia, thalamus, and cortico-white matter junction also have tightly packed grey and white fibres.59,60 Disruption of blood-brain barrier and endothelial injury leads to oedema, which may cause compression of nerve fibres, and therefore demyelination, and it can also can cause damage to myelin by releasing excitotoxins, such as glutamate. 61
In most previous studies, HbA1c has been assessed, but Maccario et al. found that serum osmolality has a more important role than hyperglycaemia or hyponatremia, because the neurological manifestations of NKH do not develop in patients with normal serum osmolality. In addition, a rapid increase in blood glucose concentration, rather than prolonged hyperglycaemia, is considered to be more important in the pathogenesis of the condition. Rapid hyperglycaemia-induced diuresis creates a sudden steep gradient between the extracellular and intracellular compartments. 3 The two cases described herein were characterised by long-term symptoms, which suggests that a longer duration of symptoms may result in more severe neurological deficits.
Homonymous hemianopsia, aphasia, hemisensory defects, hemiparesis, hyperreflexia, and the Babinski sign, along with both simple and complex visual hallucinations, all suggest the presence of diffuse cortical or subcortical damage. 1
Diagnostic investigations
Laboratory parameters
Patients with NKH have moderate-to-severe hyperglycaemia and high HbA1c levels, implying poor long-term glucose control, although an acute increase in blood glucose can be responsible for similar symptoms. In many cases, diabetes is diagnosed after the onset of these symptoms, and patient 1 was diagnosed as having diabetes mellitus after admission because of neurological symptoms. She showed a slight-to-moderate increase in serum osmolality, in the absence of circulating ketones. A mild-to-moderate electrolyte imbalance is also common, but other routinely measured biochemical and haematological parameters are usually normal, and CSF analysis does not reveal abnormalities17,62.
Electroencephalography
In contrast to the generalised seizures that categorise hypoglycemia, 63 hyperglycaemia in general causes focal seizures, 64 which may be related to the presence of K-ATP channels in certain parts of neocortex. 65 EEG generally reveals focal epileptiform activity,11,13,22,39 with sharp or spike wave activity apparent on the posterior cortical leads. Unilateral or bilateral, asynchronous or synchronous focal epileptiform discharges are most common in the occipital region, followed by the temporal and parietal regions. Patients can also develop EPC,38,39 which is often characterised by continuous focal epileptic activity on EEG, and generalised or focal slowing is common. However, patients can also display normal electroencephalograms.
Visual evoked potential
Pattern-reversal visual evoked potential has been performed in a patient with visual seizures, and this shows a large unilateral P100 amplitude.22,66
Imaging findings
Brain computed tomography
Computed tomography (CT) typically does not show any changes; therefore, a diagnosis of NKH cannot be made using CT. Indeed, corroborative CT changes have not been identified in any of the reported cases, including when contrast-enhanced CT was used7.
MRI
T1-weighted images do not show changes, but contrast-enhanced images often show a gyral or leptomeningeal enhancement pattern. T2 hypointensity in the subcortical posterior cerebral region is the most characteristic finding on MRI, and FLAIR images show similar hypointensity. In addition, contrast FLAIR has been shown to demonstrate enhancement patterns better than contrast T1. Diffusion-weighted images may or may not show restriction. 67 MR angiography does not show any stenosis or paucity of intracranial vessels. Most published cases have shown resolution of the T2 hypointensity on follow-up scans after few weeks or months, but a few have shown focal atrophy in the same region on follow-up scans. 16 A susceptibility-weighted imaging (SWI) sequence can show small hypointense foci that represent microhaemorrhages or the presence of gemistocytes. 24 In one of the present cases, there was diffusion restriction and a hypointense signal on FLAIR in the central pontine region in addition. Central pontine hyperintensity has also been reported in a patient with NKH, a hyperosmolar state, and EPC. 59 Because the present case featured hypointensity, as opposed to the pontine hyperintensity reported by Mao et al. 58 , osmosis and secondary demyelination likely played a role in the pathogenesis.
A comparison with the imaging findings that typify hyperglycaemic chorea may also be instructive. In patients with chorea, unenhanced T1-weighted images show hyperintensity in the basal ganglia region, 2 and most commonly in the putamen, followed by the caudate and globus pallidus, whereas T2-weighted images may feature hyper-,hypo-, or isointense signals. 68
Shan et al. linked a hyperintense T1 signal to a layer of hydrated proteins inside the cytoplasm, which typifies gemistocytes. Stereotactic biopsy reveals abundant gemistocytes, which are swollen reactive astrocytes with a high protein content that are typically seen after an acute injury, and subsequently shrink. 69 According to Chu et al., patients with chorea and ballismus resulting from hyperglycaemia show normal gradient echo (GRE) images, whereas diffusion-weighted imaging (DWI) shows restricted diffusion, consistent with hyperviscosity, rather than petechial haemorrhages as the cause of the cytotoxic oedema and imaging findings. 70 Nevertheless, a few authors have suggested that greater paramagnetic deposition may be the cause of putaminal hypointensity on SWI images. 71
Magnetic resonance spectroscopy (MRS)
It is advisable to perform MRS alongside routine sequences. In some previous studies, MRS showed large peaks corresponding to metabolites such as choline, myoinositol, and particularly creatine, but normal lipid, lactate, glucose, and ketone peaks, 25 which is indicative of hyperosmolality. 72 In a few previously reported cases, the N-acetylaspartate (NAA) peak was small,22,66 which is suggestive of cortical laminar necrosis and neuronal loss. A small NAA peak may also suggest that cortical laminar necrosis is the cause of the gyral enhancement on T1 contrast and also implies irreversible damage.
Functional MRI
Alessandra et al. showed a positive blood oxygen-dependent (BOLD) signal in Brodmann area 18 (the visual association area) during continuous EEG 8 using blood oxygen level-dependent contrast imaging, which utilises the magnetic properties of haemoglobin. This method is sensitive to blood flow changes induced by metabolic or neuronal activity.
Fluorodeoxyglucose-positron emission tomography
Hypermetabolism in the right occipital cortex has previously been documented in a patient with hyperglycaemia-induced hemianopia and T2 hypointensity, 72 and this had resolved 3 months later.
Single-photon emission computerised tomography
Tc99m-D,L-hexamethylene-propyleneamine oxime or I123-N-isopropyl-iodoamphetamine-single-photon emission computerised tomography shows hyperperfusion during ictal activity, as confirmed using simultaneous EEG, but hypoperfusion during the interictal phase or after the symptoms resolve.15,22,23
Differential diagnosis and management
In patients with diabetes and visual defects or partial seizures, it is important to consider NKH. Many patients with NKH who present with cortical symptoms are diagnosed as being diabetic upon admission.73,74 Thus, a diagnosis could be missed if a CT examination alone is performed, and this fails to show any abnormalities. The most characteristic MRI feature is T2 hypointensity, predominantly in the posterior cerebrum. Other causes of T2 hypointensity are early stroke, metastasis, meningitis, encephalitis, multiple sclerosis, and moyamoya disease, but these causes can be ruled out on the basis of clinical presentation, physical examination, blood glucose and HbA1c, CSF examination, and the EEG and MRI findings. 75 Early detection is the key to the resolution of symptoms and the prevention of vision loss. In addition, the immediate initiation of insulin infusion, fluid and rehydration therapy, and the correction of electrolyte abnormalities are important.
Discussion
It can be inferred from the literature that abnormal circulating glucose concentrations, whether hypoglycaemia or hyperglycaemia, tend to have effects on the posterior cerebral region, and especially on the parieto-occipital region. 76 Metabolic derangement leading to a GABA shunt or the activation of kATP channels in the posterior neocortex might also be responsible for focal or occipital seizures.
Diffusion restriction can be caused by hyperviscosity, because it also characterises hyperglycaemia-induced hemichorea hemiballismus. 69 Nevertheless, plain T1-weighted images do not show changes in the posterior cerebral region resulting from hyperglycaemia, in contrast to the hyperintense signal in the basal ganglia that typifies hemichorea hemiballismus. In addition, the findings of T2 and FLAIR are dissimilar in these two conditions, which suggests differing pathophysiology, even though the aetiology of both conditions is NKH.
Although previous case reports have described only transient symptoms associated with posterior cerebral defects, both of the present cases featured irreversible vision loss, which can be explained by the identification of cortical laminar necrosis on the initial scan and focal gliosis on follow-up imaging. The presence of cortical laminar necrosis is consistent with the low NAA concentration identified using spectroscopy and the gyral enhancement pattern on contrast MRI. One of the most important reasons for irreversible visual loss is likely to have been late presentation, implying long-term metabolic derangement and neuronal hyperexcitability. The only predictor of seizure control in patients with NKH identified to date is the frequency of seizures. 77 The patients reported herein had been experiencing occipital seizures over a long period of time, which is likely to explain the irreversible loss of vision.
A diagnosis of cortical lesions secondary to NKH may be delayed because of a lack of awareness of the various manifestations, and may lead to inadequate management and a lack of full recovery. Therefore, it is important to perform a detailed clinical evaluation and brain MRI in patients with an abnormal blood glucose concentration and visual or other cortical symptoms.
It is possible that many of the cases reported previously might have had some irreversible cortical vision loss or field defects. However, because visual field testing was not performed during the monitoring of these patients, it is quite likely that mild-to-moderate unilateral field defects were missed.
In summary, the symptoms of NKH are referable to the posterior cerebral cortex. Various types of focal seizures can be seen clinically, and occipital seizures and various visual defects are the most common symptoms, with irreversible vision loss being possible. The disease is characterised by specific MRI findings, and the differences in the MRI findings in the basal ganglia versus the posterior cerebral region suggest that the pathophysiology of the epileptic manifestations of NKH and hemichorea hemiballismus differs.
Footnotes
Declaration of conflicting interest
The author declares that there is no conflict of interest.
Ethics statement
The present study was approved by the institutional ethics committee of Sir Jamshedjee Jeejeebhoy Hospital Mumbai, Jupiter Hospital. Written informed consent was obtained from the patients for their participation in the study, and for publication of the case reports and the accompanying images.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sector.