
Abstract
Extramedullary plasmacytoma (EMP) is a rare plasma cell neoplasm, with the majority (80–90% of cases) occurring in the upper aerodigestive tract. To our best knowledge, primary EMP from renal tissues is extremely rare. Herein, the diagnosis and treatment of a refractory primary EMP with renal involvement in a 53-year-old male patient is reported. The patient received radical nephrectomy followed by radiotherapy, and showed relapse 3 months after treatment. The cancer cells were sensitive to subsequent chemotherapy, however, the patient died of infection associated with the disease after almost 3.5 years following first presentation.
Introduction
Extramedullary plasmacytoma (EMP) is a rare plasma cell neoplasm that occurs in the soft tissues without bone marrow involvement. 1 The majority of EMP lesions are localized to the head and neck, with rare cases presenting primary EMP in the kidney. 2 Here, a case of primary renal plasmacytoma, confirmed by histopathological and immunohistochemical examinations, is presented, together with a review of the literature regarding renal plasmacytoma.
Case report
Publication of this case report was approved by the Ethics Committee of Binzhou Medical University Hospital, China (No. 2021-LW-018). The reporting of this study conforms to CARE guidelines, 3 and written informed consent for treatment and publication of the case was obtained from the patient.
A 53-year-old male presented at the Department of Urology, Binzhou Medical University Hospital, in December 2016 with a 12-month history of frequent and painful micturition. Abdominal ultrasonography indicated a mass in the left kidney. Physical examination findings were normal, and medical laboratory tests showed normal ranges. Serum lactate dehydrogenase was 160.7 U/L (normal range, 109–245 U/L), and beta-2 microglobulin was 5.4 μg/ml (normal range, 3.2–6.5 μg/ml). Chest computed tomography (CT) and radionuclide bone imaging findings showed no abnormalities. Contrast-enhanced CT of the kidney revealed a large mass (12 × 8 × 6 cm) with heterogeneous enhancement in the upper pole of the left kidney, and partial lesions adjacent to the renal artery and vein. No enlarged lymph nodes were identified in the retroperitoneal region; however, a suspicious metastasis (diameter, 1.8 cm) was detected in the right anterior lobe of the liver. The patient reported no previous history of renal diseases, and was initially diagnosed with renal cell carcinoma (RCC; Figure 1).
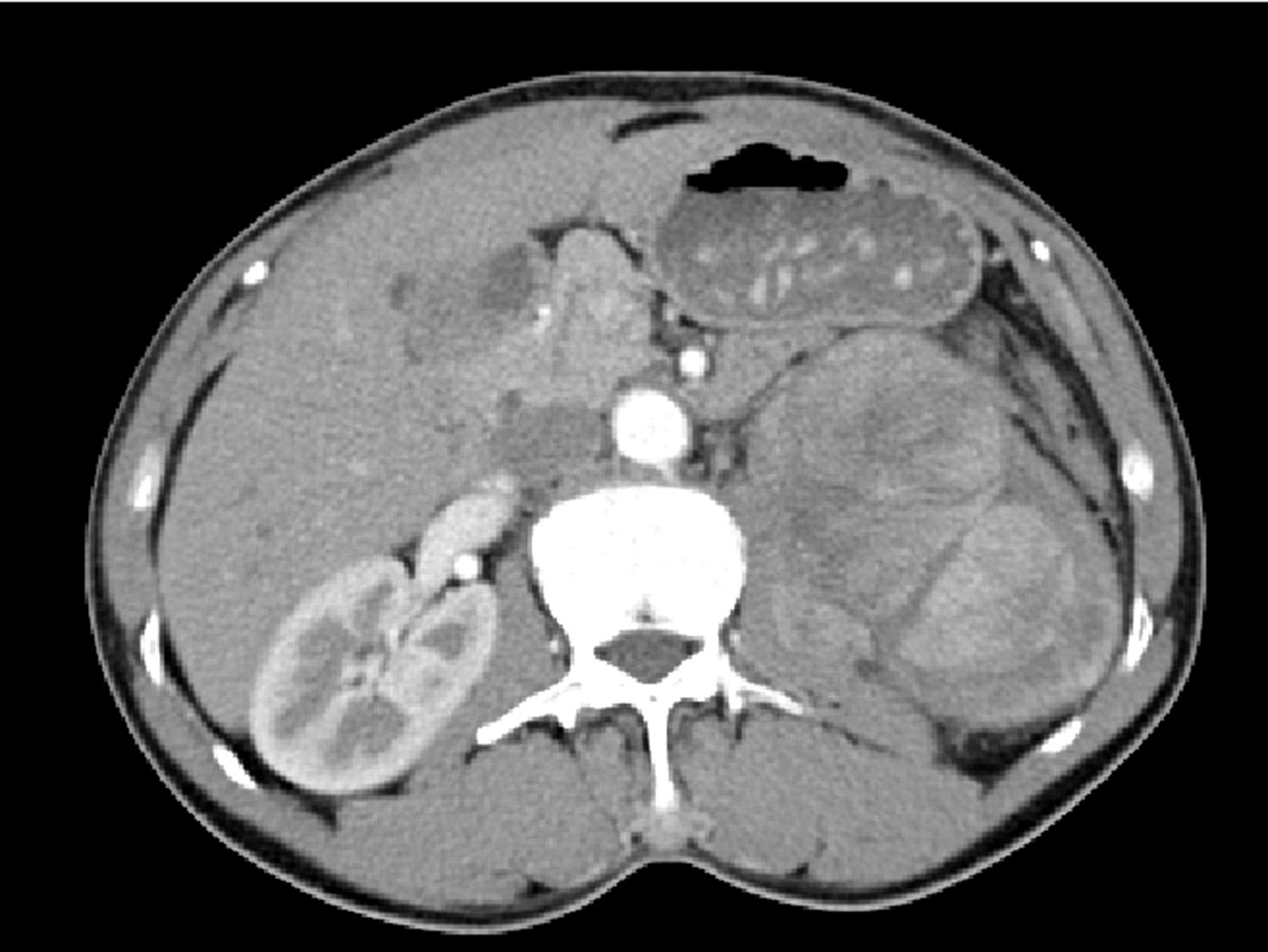
Contrast-enhanced abdominal computed tomography transverse plane image from a 53-year-old male patient, showing a large mass with heterogeneous enhancement in the upper pole of the left kidney.
Prior to treatment, the patient was advised to undergo core biopsies of the renal and hepatic masses, however, he requested direct surgical treatment. The patient had a Karnofsky Performance Status (KPS) score of 90% (KPS score range: 0–100, where 100% equates to ‘normal no complaints; no evidence of disease’) and only one site of hepatic metastasis was suspected. Therefore, the patient received radical nephrectomy under general anaesthesia in December 2016. The mass was located in the upper renal lobe and was firmly anchored to the renal tissues. Under gross inspection, the tumour was found to be an unencapsulated mass (11 × 7 × 6 cm) with clear margins and of a faint yellow colour. There were no enlarged lymph nodes surrounding the kidney.
Histological analysis of the resected tumour showed infiltration with plasmacytoid cells of various sizes and levels of differentiation (Figure 2A and B). Immunohistochemical analysis demonstrated positive immunostaining for CD38 (antibody [Ab] clone SPC32), CD138 antigen (Ab clone EP201), and lambda light chain (Ab clone SHL53) (Figure 2C–E), together with apoptosis regulator Bcl-2 (Ab clone SP66), vimentin (Ab clone MX034) and multiple myeloma oncogene 1 (MUM-1; Ab clone EP190). In addition, a proportion of the cells were positive for CD79a antigen (Ab clone SP18), cyclin D1 (Ab clone EP12) and proto-oncogene c-Myc (Ab clone EP121). The cancer cells were negative for kappa light chains (Ab clone CH15), B-lymphocyte antigen CD20 (Ab clone L26), CD3 (Ab clone UMAB54), CD5 antigen (Ab clone UMAB9), membrane metalloendopeptidase (CD10; Ab clone UMAB235), B-cell lymphoma 6 protein (Ab clone LN22), myeloperoxidase, cytokeratin, CD45 antigen (LCA; Ab clones 2B11 & PD7/26), CK-7 (Ab clone OV-TL12130), S-100 protein, myogenic differentiation 1 (Ab clone EP212) and myogenin (Ab clone EP162). The Ki-67 index was found to be approximately 30–40% (Figure 2F).

Representative photomicrographs of resected kidney tumour tissue from a 53-year-old male patient showing: (a) massive proliferation of mononuclear cells infiltrating the kidney (haematoxylin and eosin [H&E] staining; original magnification, ×20); (b) differences in the size and degree of differentiation between plasmacytoid cells (H&E staining; original magnification, × 400); (c) CD38-positive immunostaining (original magnification, ×200); (d) CD138-positive immunostaining (original magnification, ×200); (e) lambda light chain (antibody clone SHL53)-positive immunostaining (original magnification, ×200); and (f) Ki-67-positive nuclear immunostaining with a Ki67 index of 30–40% (original magnification, ×100).
A skeletal survey showed no lytic bone lesions or evidence of active malignancies elsewhere, and bone marrow aspirate was normal. Urinalysis for Bence-Jones protein was negative and serum protein electrophoresis excluded multiple myeloma. There was no evidence of systemic plasma cell disease, thus, renal EMP of a solid nature was proposed (tumour stage II or more).
Extramedullary plasmacytoma is highly sensitive to radiotherapy. 2 The patient received intensity-modulated radiation therapy (T11-L4) at a dose of 50 Gy (22 fractions, 38 days) from January 2017. In May 2017, abdominal CT showed metastatic lesions in the right adrenal gland, kidney and muscle groups at the vertebral side. Chest CT and abdominal enhanced CT indicated additional nodules in the cardiac apex, the left lateral lobe of the liver, and the levator ani muscle. The patient developed progressive disease, however, a repeated bone marrow aspiration and immunofixation electrophoresis for immunoglobulin (Ig)G, IgA, IgM, κ, and λ, did not return positive results. The patient received one cycle of chemotherapy comprising 0.4 g cyclophosphamide, intravenously (i.v.), once daily (day 1, 10 and 15); 10 mg pirarubicin, i.v., once daily (day 1–4); 40 mg dexamethasone, orally, once daily (day 1–4 and day 11–14); and 100 mg thalidomide, i.v., once daily (day 1–15). An additional two cycles of chemotherapy were then administered using an IADP regimen comprising 2 g ifosfamide, i.v., once daily (day 1–3); 20 mg cisplatin, i.v., once daily (day 1–4); 40 mg pirarubicin, i.v., once daily (day 1); and 20 mg dexamethasone, orally, once daily (day 1–4 and day 9–12).
In August 2017, chest and abdominal CT revealed nodules under the left abdominal wall that were reduced in size, indicating partial response to treatment. Consequently, the patient received a further four cycles of chemotherapy using the IADP regimen and treatment response was classified as morphological complete remission. Positron emission tomography/CT, performed in June 2018, revealed new neoplasms in the retroperitoneum, pelvic cavity and right thigh. There was no evidence of a plasma cell neoplasm in the bone marrow. The patient received three cycles of IADP chemotherapy, however, chest and abdominal CT revealed no response. Further chemotherapy comprising one cycle of GEP (1.4 g gemcitabine, i.v., once daily, day 1 and day 8; 0.1 g etoposide, i.v., once daily, day 1–4; and 20 mg dexamethasone, orally, once daily, day 1–4), also showed no response. Fine needle biopsy of the femoral lesion, performed in January 2019, demonstrated plasma cell infiltration, and the morphology was in line with renal plasmacytoid cells. The patient received radiotherapy in March 2019 for the right femoral lesion, at a dose of 50 Gy (20 fractions, 32 days). Flow cytometry of repeated bone marrow aspirate indicated plasma cells (0.27%). The patient subsequently received one cycle of chemotherapy comprising 25 mg lenalidomide, i.v., once daily (day 1–21); 0.8 g cyclophosphamide, i.v., once daily (day 1); and 20 mg dexamethasone, orally, once daily (day 1–4). The patient died due to infection in May 2020 without further therapy.
Discussion
Plasmacytoma originates from the monoclonal malignant transformation of plasma cells. Solitary plasmacytoma is an early-stage plasma cell malignancy, defined by the presence of a single biopsy-proven plasmacytoma (bony or extramedullary) and a normal bone marrow examination. 2 The occurrence of EMP in the kidney is extremely rare, as most cases show involvement in the head and neck, the upper respiratory system, and the gastrointestinal tract. 2
A comprehensive literature search of the PubMed, Medline and Embase databases, for English language articles on renal plasmacytoma (excluding articles involving secondary EMPs and cases with bone marrow involvement) revealed 16 published cases (11 male and 5 female; median age, 56.8 years [range, 14–76 years]) arising in the kidney (Table 1).4–19 Among these cases, 10 (62.5%) showed primary EMP lesions in the left kidney, five (31.25%) were in the right kidney, and one (6.25%) was in both kidneys, resulting in renal insufficiency without obvious obstruction or hydronephrosis. Five cases (31.25%) presented with abdominal or back pain, and four (25%) showed haematuria. Routine laboratory tests showed no abnormalities. To our best knowledge, radiological examinations are not adequate in distinguishing RCC. Tumour size varied between 3 and 21 cm (mean, 8.7 cm). Thirteen patients received radical or partial nephrectomy, of whom, five received radiotherapy after surgery,4–8 and one received chemotherapy. 9 Two patients received chemotherapy only,10,11 and one patient underwent laparotomy and biopsies only. 12 During the follow-up period, treatment was terminated in three cases;9,13,14 and four patients died,8,11,12,15 including one from pneumonia two months after surgery, one from high tumour burden 14 years after surgery due to refractory to chemotherapy and radiotherapy, one from bronchopneumonia after laparotomy, and one from acute myocardial infarction in the 16th year following surgery. Of the other nine cases, one showed relapse 6 months after surgery and presented local symptoms shortly after metastasectomy, 6 and eight were still alive, including three patients who were followed-up for more than 1 year. Six out of the 16 patients showed comorbidities, including three with malignant tumours,5,10,13 one with hepatitis B virus and HIV infection, 4 one with hepatitis C virus infection, 6 and one with Henoch-Schönlein purpura. 16 These data suggest that chronic antigen stimulation and immunologic alterations may serve as driving forces for the progression of plasmacytoma. 20 One patient presented with collision tumour histology between EMP and clear cell RCC. 17
Summary of the present case and 16 published cases of primary renal extramedullary plasmacytoma.

F, female; M, male; L, left kidney; R, right kidney; NA, not available; HCV, hepatitis C virus; Ig, immunoglobulin; SPE, serum protein electrophoresis; IE, immunoelectrophoresis; chemo, chemotherapy.
*Bortezomib, cyclophosphamide and dexamethasone; #regimen details not available; &vincristine, adriablastin, dexamethasone, bortezomib and lenalidomide.
As renal plasmacytoma is extremely rare, it brings diagnostic challenges in clinical settings due to its location and non-specific symptoms. The diagnosis of EMP is complex and usually requires radiological, haematological, biochemical and histological examinations. Upon histological diagnosis, further systematic investigations should be conducted to exclude the possibility of multiple myeloma. Current diagnostic criteria for solitary EMP are as follows: biopsy-proven solitary lesion of bone or soft tissue with evidence of clonal plasma cells, normal bone marrow with no evidence of clonal plasma cells, normal skeletal survey and magnetic resonance imaging (or CT) of the spine and pelvis (except for the primary solitary lesion), and absence of end-organ damage, such as CRAB features (hypercalcaemia, renal failure, anaemia, and bone lesions) that can be attributed to a lympho-plasma cell proliferative disorder. 1 The patient in the present case was initially clinically misdiagnosed with RCC. After histopathological examination, immunohistochemical detection and systemic examination, a diagnosis of renal plasmacytoma was confirmed. With no common risk factors between RCC and plasmacytoma, their relationship in terms of origin and propagation is merely speculative. Ojha et al. 21 found patients with a primary RCC showed a higher risk of developing multiple myeloma, while those with primary multiple myeloma had a higher risk of developing RCC. Authors of another study speculated that interleukin-6 may be a potential mediator, playing significant roles in the pathogenesis of RCC and plasmacytoma. 17
There are currently no standard guidelines for renal plasmacytoma, and treatment options include surgery, radiotherapy and chemotherapy, alone or in combination. EMP is widely acknowledged to be highly sensitive to radiotherapy, with nearly all patients achieving local control following treatment,22,23 and a radiation dose of 40–50 Gy has been shown to contribute to local control without severe radiation injury.24,25 In some studies, radiotherapy has even been recommended for patients with complete remission presenting negative margins.26,27 Compared with surgery, radiotherapy has been associated with favourable outcomes in terms of multiple myeloma-free survival and progression-free survival. 28 In some cases, chemotherapy was advisable for those with refractory conditions or relapse, 2 and some studies have shown that initial chemotherapy is effective. 10 According to a previous report, 29 local regression did not portend a poor prognosis. The patient in the present case was at a tumour stage of II or more. He received radiotherapy following surgery, but the disease showed progression, indicating that the tumour cells were not sensitive to radiotherapy. The patient consequently received chemotherapy, and due to the refractory condition of the disease, the chemotherapeutic regimens were changed at cycle 2, resulting in a complete response. We speculate that patients with EMP and no metastasis may benefit from radiotherapy, while for those with metastasis, chemotherapy may be more beneficial. Unfortunately, the patient was not sensitive to initial treatment and showed recurrence approximately ten months later. Although the chemotherapy regimens were altered, for example to a GEP regime (which is effective for non-small cell lung cancer), or combined with lenalidomide, neither was effective. The patient declined further treatment, including stem cell transplantation.
Patients with EMP are likely to show good prognosis, with a five-year survival rate of approximately 90%. Nevertheless, more than 30% patients may present local recurrence and metastasis, and some convert to multiple myeloma. 30 EMP and multiple myeloma are two diseases with different biological behaviours. For patients with EMP who progress to multiple myeloma, chemotherapy may delay the time to progression, but a previous study showed no significant difference in median survival time between patients treated with or without chemotherapy. 30 Several prognostic factors are reported to be associated with EMP outcomes, including age, tumour size, serum M protein and treatment regimens.28,31,32 Unlike EMP in the head or neck regions, primary renal plasmacytomas are often detected late with a larger tumour size, and the prognosis is not comparable. In the present case, the patient showed recurrence and multiple metastases, and his disease was refractory to common chemotherapy regimens used to treat multiple myeloma.
The present study may be limited by the fact that the right anterior lobe lesion of the liver was not pathologically diagnosed because the lesion was cured following chemotherapy. On this basis, the tumour stage cannot be confirmed exactly.
In summary, primary renal plasmacytoma is a rare plasma cell disorder, which may present a good prognosis after treating with a combination of radiotherapy and surgery. Chemotherapy is an optional approach for refractory cases. Patients with primary renal plasmacytomas require long-term follow-up and close monitoring.
Supplemental Material
sj-pdf-1-imr-10.1177_03000605211063713 - Supplemental material for Refractory primary extramedullary plasmacytoma in kidney: a case report
Supplemental material, sj-pdf-1-imr-10.1177_03000605211063713 for Refractory primary extramedullary plasmacytoma in kidney: a case report by Wenjie Niu, Lili Zhang, Yuhai Wu, Kai Li, Lixia Sun, Hong Ji and Bing Zhang in Journal of International Medical Research
Footnotes
Data accessibility
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and publication of this article: This work was funded by grants from Medical and Health Science and Technology Development Project of Shandong Province [Grant No. 2018WS547, to Bing Zhang; Grant No. 2015WS0494, to Hong Ji]; and the Natural Science Foundation of Shandong Province [Grant No. ZR2020MC068, to Hong Ji].
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.