
Abstract
Objective
Glioblastoma (GB) is a refractory malignancy with a high rate of recurrence and treatment resistance. Hypoxia-related genes are promising prognostic indicators for GB, so we herein developed a reliable hypoxia-related gene risk scoring model to predict the prognosis of patients with GB.
Method
Gene expression profiles and corresponding clinicopathological features of patients with GB were obtained from the Cancer Genome Atlas (TCGA; n = 160) and Gene Expression Omnibus (GEO) GSE7696 (n = 80) databases. Univariate and multivariate Cox regression analyses of differentially expressed hypoxia-related genes were performed using R 3.5.1 software.
Result
Fourteen prognosis-related genes were identified and used to construct a risk signature. Patients with high-risk scores had significantly lower overall survival (OS) than those with low-risk scores. The median risk score was used as a critical value and for OS prediction in an independent external verification GSE7696 cohort. Risk score was not significantly affected by clinical-related factors. We also developed a prediction nomogram based on the TCGA training set to predict survival rates, and included six independent prognostic parameters in the TCGA prediction model.
Conclusion
We determined a reliable hypoxia-related gene risk scoring model for predicting the prognosis of patients with GB.
Introduction
Glioblastoma (GB) is a refractory malignant tumor which is prone to relapse and resistant to treatment. 1 Despite considerable advances in GB therapies, the optimal treatment strategy remains controversial. 2 Moreover, several clinical treatments for GB have been attempted during the past decade, but results are unsatisfactory, 3 , 4 and 5-year overall survival (OS) rates are about 5%.
Developments in molecular biology, including the release of the human glioma cancer gene profile, have enabled the underlying mechanisms of GB to start to be elucidated. 5 Several molecular mechanisms and signal transduction pathways have been confirmed to play a key role in GB carcinogenesis, and their in-depth analyses are important in improving the prognosis of patients with GB.6,7 Caspases, B-cell lymphoma-2, members of the inhibitor of apoptosis family, and p53 have been identified as potential drug targets for GB treatment. 8 Furthermore, targeted interventions may be possible for GB subsets including BRAF mutations, fusion of neurotrophic tryptophan receptor kinase or fibroblast growth factor receptor genes, and c-Met encoding gene amplifications or fusions. 9
Hypoxia is a common microenvironment for solid tumors that can occur under both physiological and pathological conditions. It has a great impact on tumor metastasis, and radiotherapy and chemotherapy sensitivity, and is associated with a poor prognosis. 10 Vasculogenic mimicry (VM) is a functional microcirculation pattern formed by most solid tumors to adapt to hypoxia. VMs are present in cervical cancer, prostate cancer, osteosarcoma, and other malignant tumors. Hypoxia also promotes brain tumorigenesis, 11 so hypoxia-related genes are promising prognostic indicators in GB. Indeed, some studies have shown that hypoxia is the driving force of GB and could be used as a novel treatment tool. 12 Other studies reported that the inhibition of hypoxia-mediated mitochondrial apoptosis induces temozolomide resistance through the microRNA-26a/Bad/Bax axis. 13 However, there is currently no prognosis model for GB constructed by hypoxia-related genes. Therefore, in this study, we determined a reliable hypoxia-related gene risk scoring model to predict prognosis in patients with GB.
Methods
Ethical approval
This study did not require approval by the Ethics Committee because it was based on data from a public database.
Identification and enrichment analysis of differentially expressed hypoxia-related genes
The edgeR package from R 3.5.1 software (www.r-project.org) was used to screen differentially expressed hypoxia-related genes (HDEGs) in normal samples from the Cancer Genome Atlas (TCGA) GB cohorts. Adjusted P < 0.05 and | (fold-change) | >1.5 were considered significant in identifying DEGs. DEGs in the TCGA cohort were visualized as volcano plots. Heatmaps were constructed using the “heatmap” R package. A complete and up-to-date list of human genes directly associated with hypoxia was obtained from the Gene Ontology (GO) resource database. Functional annotation and pathway enrichment analyses were performed using GO and Kyoto Encyclopedia of Genes and Genomes (KEGG). A P-value less than 0.05 was considered statistically significant.
Gene set enrichment analysis
Gene expression levels were set to the population phenotype, and GSEA was used to evaluate related pathways and molecular mechanisms in patients with GB. Enriched gene sets with a P value less than 0.05 and a false discovery rate less than 0.25 were considered statistically significant.
Development of the prognostic risk scoring model
HDEGs associated with the prognosis of GB were identified by univariate and multivariate regression analyses. The risk model was constructed using HDEG derived from multivariate regression analysis. The correlation coefficient (CC) of each gene was obtained by multi-factor analysis, and the risk score was calculated as follows: ADM×CC1+PML×CC2+SUV39H2×CC3+PLOD2×CC4+PSMC2×CC5+AQP3×CC6+CAV1×CC7+CYGB×CC8+MMP14×CC9+MMP2×CC10+PAK1×CC11+PHB2×CC12+HSPBAP1×CC13+SLC9A1×CC14.
Patients were divided into high-risk and low-risk groups according to the median risk score. The performance of the hypoxia-associated differential gene (HATG)-based risk scoring model constructed from the TCGA training set was verified by a similar method using GEO cohorts. Clinical data were used to construct a nomogram of the clinical survival prediction model based on six independent prognostic parameters (age, sex, ethnicity, radiation therapy, chemotherapy, and risk score) using the rms package in R.
Statistical analysis
For descriptive statistics, means ± standard deviations were used for continuous variables in a normal distribution, while medians (ranges) were used for continuous variables in non-normal distributions. Categorical variables were described by counts and percentages. Two-tailed p < 0.05 was regarded as statistically significant.
Results
A total of 203 HDEGs were identified in the TCGA dataset, as shown in the heatmap and volcano plot (Figure 1a and 1b). GO analysis showed that HDEGs were significantly enriched in threonine-type endopeptidase activity (Figure 1c) in the molecular function category. KEGG showed that HDEGs were also mainly enriched in the hypoxia-inducible factor (HIF)-1 signaling pathway (Figure 1d).
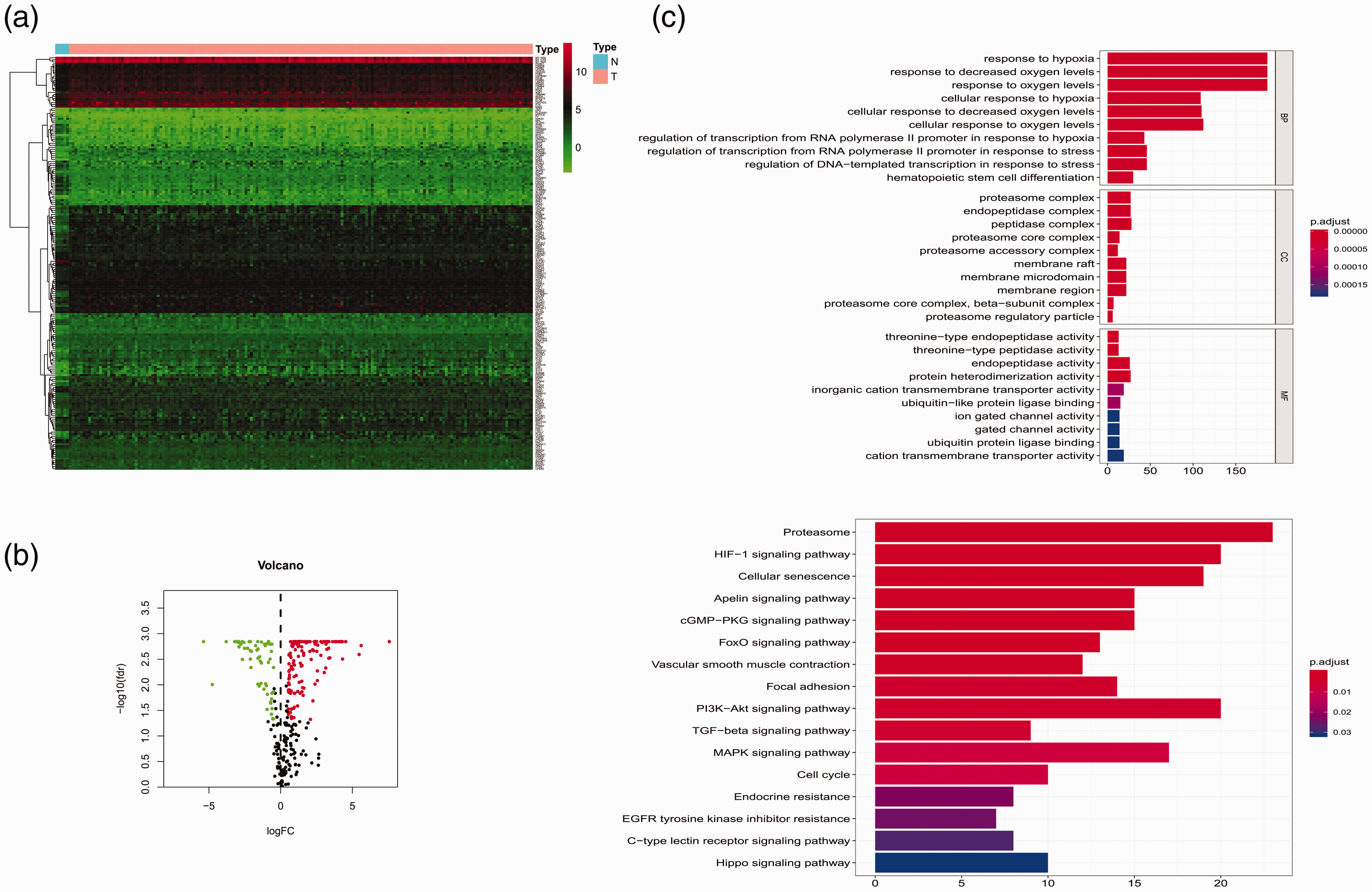
Differentially expressed hypoxia-related genes and the identification of hub hypoxia-related genes. (a) Heatmap of differentially expressed hypoxia-related genes in the TCGA. (b) Volcano plot of differentially expressed hypoxia-related genes in the TCGA. (c) GO and (d) KEGG for differentially expressed hypoxia-related genes.
Fourteen prognosis-related genes with significant prognostic value (P < 0.05) were identified: ADM, CAV1, PHB2, PLOD2, HSPBAP1, PSMC2, SUV39H2, MMP14, PML, PAK1, MMP2, SLC9A1, CYGB, and AQP3.
The prognostic risk scoring model was developed from the following formula: Risk score = ADM×0.39558447+PML×(−0.319838772)+SUV39H2 × 0.082177321+PLOD2×(−0.073192408)+PSMC2 × 0.070433769+AQP3×(−1.199378861)+CAV1×(−0.551480788)+CYGB×0.498531769+MMP14 × 0.164031848+MMP2 × 0.078937358+PAK1 × 0.306090337+PHB2×(−0.303970421)+HSPBAP1 × 0.389250763+SLC9A1 × 1.514754259.
Subsequently, the prognostic risk score of each patient in the TCGA training set was calculated. Patients with high-risk scores had significantly lower OS (P < 0.001) than those with low-risk scores. The median risk score (−2.53) was used as a critical value and for OS prediction in an independent external verification GSE7696 cohort (P < 0.001) (Figure 2). The risk score was shown to be a significant independent prognostic factor (P < 0.05, Figure 3), and not to be affected by clinical factors.
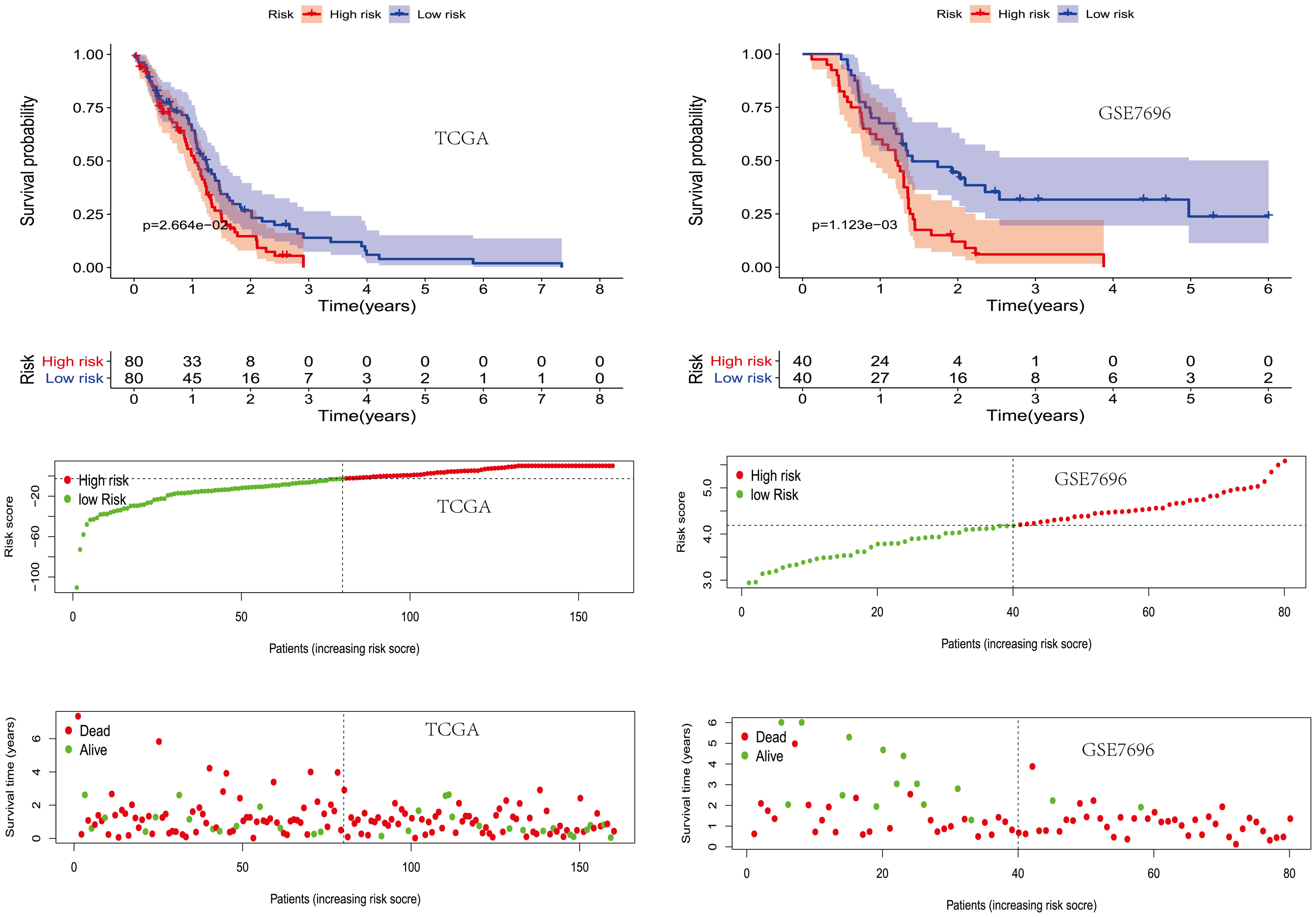
The prognostic value of the hypoxia-related risk score in TCGA and GSE7696 datasets.

Single factor and multi-factor analyses of TCGA and GSE7696 datasets.
Development and verification of nomograms
We developed a prediction nomogram based on the TCGA training set to predict survival rates. Six independent prognostic parameters were included in the prediction model. Area under the curve (AUC) values of risk scores predicting prognosis were acceptable. The nomogram also showed a good AUC in TCGA and GSE7696 databases, which verified its reliability (Figure 4).

Development of the nomogram based on hypoxia-related genes and clinical features.
GSEA analysis
GSEA analysis revealed that the high-risk group in the TCGA GB cohort was mainly enriched in the Notch signaling pathway (Figure 5).

GSEA analysis of the high-risk group in the TCGA GB cohort.
Discussion
GB displays high cellular heterogeneity, and different GB samples have different pathological and molecular characteristics. Therefore, the discovery of effective individualized treatment targets and the refining of prognosis evaluation are of great importance for patients with GB. Multiple gene expression patterns have previously been investigated in GB, but no prognostic model based on hypoxia-related genes has yet been developed. In the present study, we first identified 203 HATGs based on the TCGA database, then showed that 14 of these were significantly associated with prognosis. Finally, we developed and verified a novel GB prognostic signature based on HATG expression.
Several studies have previously reported a correlation between some of these prognosis-related genes and cancer. For example, jumonji domain-containing 1A, lysin-9 of di-methylated histone H3, and adrenomeduline were shown to be predictors of progression, as well as possible therapeutic targets, for patients with oral and oropharyngeal cancer. 14 E3 ligase E6-associated protein and promyelocytic leukemia were identified as potential prognostic markers in localized prostate carcinoma, 15 suppressor of variegation 3-9 homolog 2 promoted colorectal cancer proliferation and metastasis via trimethylation of the SLIT1 promoter, 16 while hypoxia-inducible factor-1-regulated procollagen-lysine, 2-oxoglutarate 5-dioxygenase 2 was a potential regulator of peritoneal gastric cancer metastasis. 17 Moreover, proteasome 26S subunit ATPase 2 promoted cancer cell proliferation, 18 aquaporin (AQP)3 and AQP5 co-expression in esophageal squamous cell carcinoma correlated with aggressive tumor progression and poor prognosis, 19 while caveolin 1 promoted hepatocellular carcinoma cell progression, 20 and globin family member cytoglobin suppressed breast cancer progression by inhibiting glucose metabolism. 21 Additionally, matrix metalloproteinase 14 predicted poor prognosis in patients with colorectal cancer, 22 PAK1 was an important oncogene in non-small cell lung cancer, 23 prohibitin 2 promoted cancer cell migration by inhibiting AKT serine/threonine kinase 2 expression, 24 and heat shock 27 kDa)-associated protein 1 facilitated prostate cancer cell growth. 25 Solute carrier family 9A1 was also identified as a marker of glioma tumorigenesis and prognosis. 26
Hypoxia is essential for the development of gliomas and, as well as reducing the efficacy of radiotherapy, induces and promotes the proliferation and invasion of cancer cells. 27 Si306 is a new candidate GB drug that was reported to be effective against glioma resistance to conventional therapy. Studies have also shown that under the action of hypoxia and physical oxidation, metformin reduces the oxygen consumption rate of GB cell lines and partially reverses changes to hypoxia-related genes, thereby regulating the growth of cancer cells. 28 Genetic or pharmacological inhibition of branched-chain amino acid transferase 1 has also been shown to inhibit the growth of GB multiforme cells under hypoxic conditions. 29
In support of this, our results showed that hypoxia-related genes are closely related to the prognosis of patients with GB. Based on HATG expression, we developed and verified a novel prognostic signature. Compared with clinical risk factors, the HDEG-based prognostic risk scoring model score improved the capacity to predict the survival of patients with GB. This has the potential to enable more personalized treatment strategies to be developed for high-risk patients, which could include more aggressive treatments and closer follow-ups to detect relapses as soon as possible.
There are some limitations to this study. First, detailed information about neuroimaging and resection was not included. Second, our prediction model should be further validated in multi-center studies.
In conclusion, we developed a reliable hypoxia-related gene risk scoring model to predict prognosis in patients with GB. Further large-scale, multi-center research should be performed in prospective clinical cohorts to validate the prognostic model developed in this study.
Footnotes
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Funding
The authors received the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Natural Science Foundation of China (Grant no. 81671819 [LKC]).