
Abstract
Objective
In this study, we aimed to identify prognostic immune-related genes and establish a prognostic model for laryngeal cancer based on these genes.
Methods
Transcriptome profiles and clinical data of patients with laryngeal cancer were downloaded from The Cancer Genome Atlas database. Integrated bioinformatics analyses were performed to identify genes associated with prognosis.
Results
Thirty prognostic immune-related genes for laryngeal cancer were identified. We constructed a regulatory network of prognosis comprising transcription factors and immune-related genes. Multivariate Cox regression analyses identified 15 immune-related genes in the network that were used to establish the prognostic model. The model exhibited excellent prognostic prediction ability with a high area under the curve value (0.916). The calculated risk score based on expression of the 15 immune-related genes was shown to be an independent prognostic factor for laryngeal cancer.
Conclusion
We identified prognostic immune-related genes and established a prognostic model for laryngeal cancer, which might help identify novel predictive biomarkers and therapeutic targets of laryngeal cancer.
Introduction
Laryngeal cancer originates in the epithelial tissue of the laryngeal mucosa. Morbidity and mortality rates associated with laryngeal cancer are increasing worldwide, 1 , 2 and currently, there is no definite method for diagnosing laryngeal cancer. Furthermore, patients with laryngeal cancer have high rates of recurrence and often develop resistance to chemotherapy or radiotherapy. Therefore, the prognosis for patients with advanced laryngeal cancer remains unsatisfactory, 3 , 4 and more effective predictive and therapeutic targets are required.
At present, immunotherapy is one of the most promising cancer treatments. 5 Immunotherapy is based on blocking inhibitory immune checkpoints to potentiate the immune response to cancer. 5 Immune checkpoints such as cytotoxic T-lymphocyte antigen 4 (CTLA-4)/B7 and the programmed death 1/programmed death ligand 1 (PD1/PD-L1) axes have been shown to be associated with the efficacy of treatment of melanoma, renal cell carcinoma, and non-small-cell lung cancer (NSCLC).6–8 Some studies have analyzed immune genes in laryngeal cancer, but to our knowledge, the transcription factor–immune gene regulatory networks and prognostic models have not been discussed in previous studies.
In this study, we aimed to identify immune-related genes in laryngeal cancer and to explore the relationship between these immune-related genes and the prognosis of laryngeal cancer. We hoped to identify novel predictive biomarkers and therapeutic targets of laryngeal cancer.
Materials and methods
Data acquisition
Transcriptome profiling data of laryngeal cancer were downloaded from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov). Clinical data such as age, sex, grade, stage, and survival outcome were also obtained from TCGA database. Immune-related human genes were obtained from the ImmPort database (https://www.immport.org/). Transcription factor data were downloaded from Cistrome database (https://cistrome.org/). Sample information and data used in this study were all downloaded from public databases; therefore, no patient consent or ethics committee approval was necessary.
Identification of DEGs
Differentially expressed genes (DEGs) between laryngeal cancer and non-cancerous samples were screened using the Wilcoxon rank test in R software (version 3.6.1; https://www.r-project.org/). The DEGs of immune-related genes and transcription factors were also screened. A |log2 fold change (FC)| >1 and adjusted P-value < 0.05 were considered statistically significant.
Bioinformatics analysis
In total, 111 patients were screened for analysis. The expression matrix of immune-related genes was merged with survival data to identify prognostic genes. Correlation analysis between transcription factors and prognostic-associated immune genes was performed, and Cytoscape software (version 3.7.1; https://cytoscape.org/) was used to visualize molecular interaction networks. The prognostic model was established with highly upregulated and downregulated genes based on risk score. Univariate Cox regression analyses were performed to search for prognostic-associated immune genes using the Survival package (version 3.1-7) in R, and multivariate Cox regression analysis was performed to establish a risk score. The risk score was calculated as follows: risk score = Expression gene1 × βgene1 + Expression gene2 × βgene2 + Expression gene3 × βgene3 + … + Expression gene n × βgene n , where β is the regression coefficient used as the weight. The Kaplan–Meier survival curve was based on risk scores for validation. Receiver operating characteristic (ROC) curves were used to compare the accuracy of the prognostic model, and the value of the area under the curve (AUC) was calculated using the survival ROC package (version 1.0.3) in R. The risk score map, survival status distribution map, and expression heat map were drawn using the pheatmap package (version 1.0.12) in R. The expression matrix of clinical data was merged with risk score data to perform independent prognostic analysis.
Statistical analysis
Data were analyzed using R software (https://www.r-project.org/). A P-value <0.05 was considered statistically significant. The Pearson correlation test was used to analyze correlations between molecules. The survival curves were compared using Kaplan–Meier method and log-rank test.
Results
Immune-related gene expression in laryngeal cancer
All samples were obtained from the TCGA database and 5494 laryngeal cancer DEGs were screened. Immune-related genes were downloaded from the ImmPort database, and we extracted 432 DEGs using a threshold of |log2FC| >1 and P-value < 0.05. Among the DEGs, we identified more upregulated (371) than downregulated (61) immune-related genes. The heatmap and volcano map of DEGs are shown in Figure 1.

Differentially expressed immune-related human genes in laryngeal cancer. (a) Heatmap of 432 differentially expressed immune-related genes. Genes with higher expression are shown in red, genes with lower expression are shown in green, and genes that are not differentially expressed are shown in black. The bar at the top indicates sample source: blue = non-cancerous samples; pink = tumor samples. (b) Volcano plot of differentially expressed immune-related genes
Prognostic immune-related genes in laryngeal cancer
The expression matrix of immune-related genes was merged with survival data, and univariate Cox regression was used to analyze the prognostic immune-related genes. Among the 432 DEGs, 30 immune-related genes were prognostic factors for laryngeal cancer. The resulting forest plot is shown in Figure 2.

Forest plot of univariate Cox regression analyses showing 30 immune-related genes identified as prognostic factors for laryngeal cancer. An HR value >1 (red) indicates high-risk genes predicting poor prognosis, and an HR value <1 (green) indicates low-risk genes predicting good prognosis
Molecular interactions between transcription factors and immune-related genes
Transcription factors were downloaded from the Cistrome database, and 65 differentially expressed transcription factors were extracted. The heatmap and volcano map of differentially expressed transcription factors are shown in Figure 3. Correlation analysis was performed between transcription factors and immune genes, with selection criteria as follows: |correlation coefficient| >0.4 and P-value < 0.05 (Supplemental Table S1). The molecular interaction networks indicated that the immune-related gene TFRC was positively regulated by transcription factors BRCA1, HEY1, BL1XR, TP63, LEF1, E2F7, CBX2, and the immune-related gene PPARG; immune-related genes AQP9, FPR2, L13RA2, PROK2, and AHNAK were positively regulated by transcription factor ETS1; and immune-related genes XCL2, ZAP70, TNFRSF4, LCK, TRBC1, and TRBJ2-3 were positively regulated by transcription factor BATF. However, immune-related gene AHNAK was negatively regulated by transcription factors CBX8, IRF3, and USF1, as shown in Figure 4.
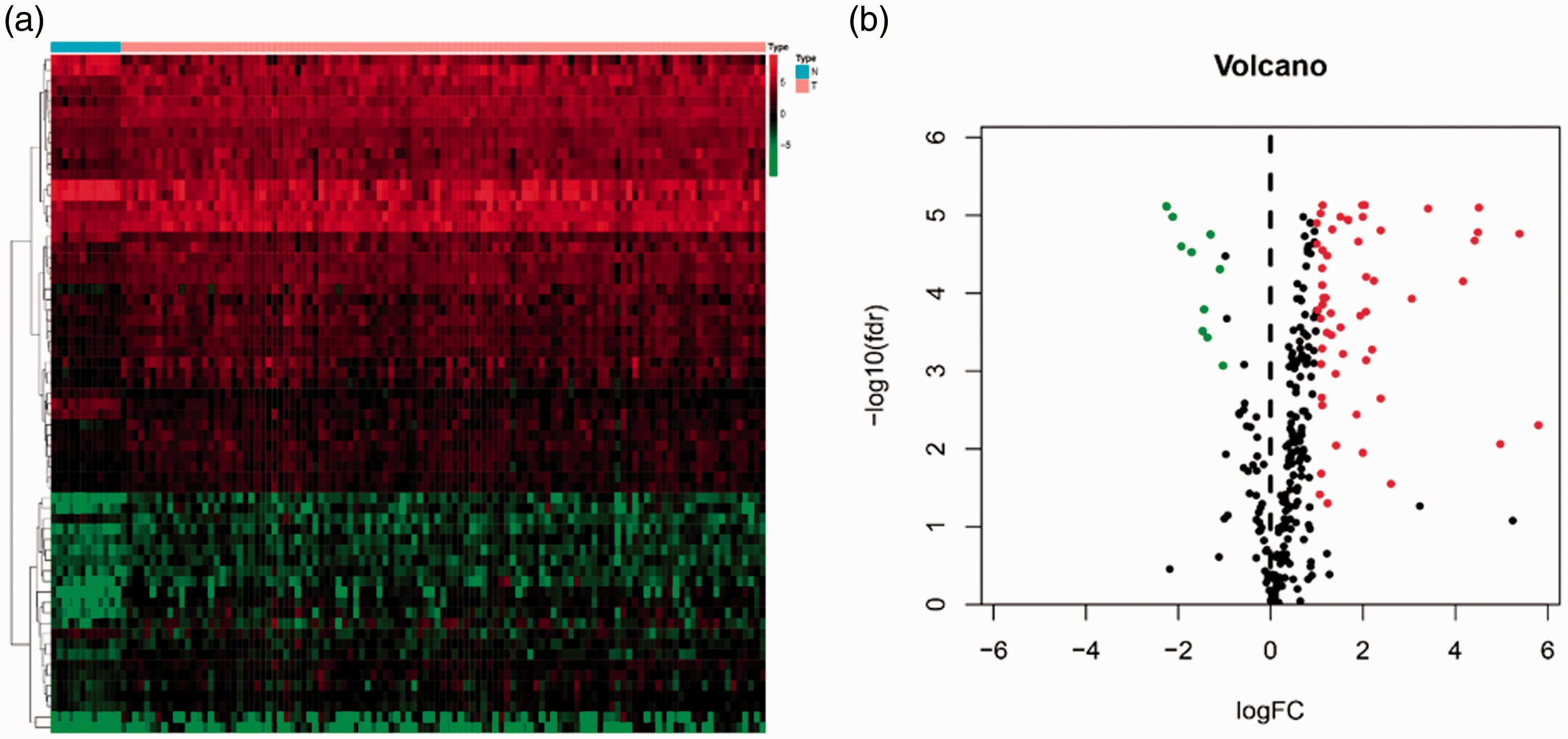
Differentially expressed transcription factors associated with laryngeal cancer. (a) Heatmap of differentially expressed transcription factors. Transcription factors with higher expression are shown in red, those with lower expression are shown in green, and transcription factors that are not differentially expressed are shown in black. The bar at the indicates sample source: blue = non-cancerous samples; pink =tumor samples. (b) Volcano plot of differentially expressed transcription factors

Networks showing molecular interactions between transcription factors and immune-related genes related to laryngeal cancer. Red circles represent high-risk immune-related genes predicting poor prognosis; blue circles represent low-risk immune-related genes predicting good prognosis; yellow circles represent transcription factors. A red line represents positive regulation, whereas a blue line represents negative regulation
Establishment and validation of prognostic model
We established a prognostic risk score model with prognostic immune-related genes. Multivariate Cox regression analysis was performed to obtain risk scores; 15 immune-related genes entered the risk score model, and of these, 11 genes were upregulated (PAEP, EPO, STC2, AQP9, TNFRSF4, RBP1, FCGR3B, FPR2, CYSLTR2, TLR2, PLCG1) and 4 were downregulated (AHNAK, PPARG, CCL2, BTC) (Supplemental Table S2). The risk score formula was as follows: risk score = ExpBTC × (0.712) + ExpFCGR3B × (0.296) + ExpPPARG × (0.249) + ExpPAEP × (0.191) + ExpAQP9 × (0.109) + ExpTLR2 × (0.070) + ExpCCL2 × (0.064) + ExpSTC2 × (0.055) + ExpRBP1 × (0.015) + ExpAHNAK × (0.009) +ExpTNFRSF4 × (−0.164) + ExpPLCG1× (−0.286) + ExpFPR2 × (−0.740) + ExpEPO × (−0.896) + ExpCYSLTR2 × (−1.286), where Exp is gene expression.
A survival curve based on risk scores was drawn for validation. In accordance with the median value of the risk score, laryngeal cancer patients were divided into a high-risk group and a low-risk group, and we found that the overall survival of patients in the low-risk group was much longer than that in the high-risk group. ROC curves were also applied to compare the efficiency of the prognostic model; the AUC of the ROC was 0.916, suggesting that the prognostic model showed excellent ability to distinguish satisfactory from poor survival in patients with laryngeal cancer. The results of survival and ROC curves are shown in Figure 5. The prognostic risk score model analysis was based on risk scores and survival outcomes; consistently, we found that the overall survival of patients in the low-risk group was much longer than that in the high-risk group, as shown in Figure 6.

Kaplan–Meier survival and ROC curves used to predict survival in patients with laryngeal cancer. (a) Kaplan–Meier survival curve. Patients were divided into high risk (red line) and low risk (blue line) based on their risk scores. (b) ROC curve and AUC score
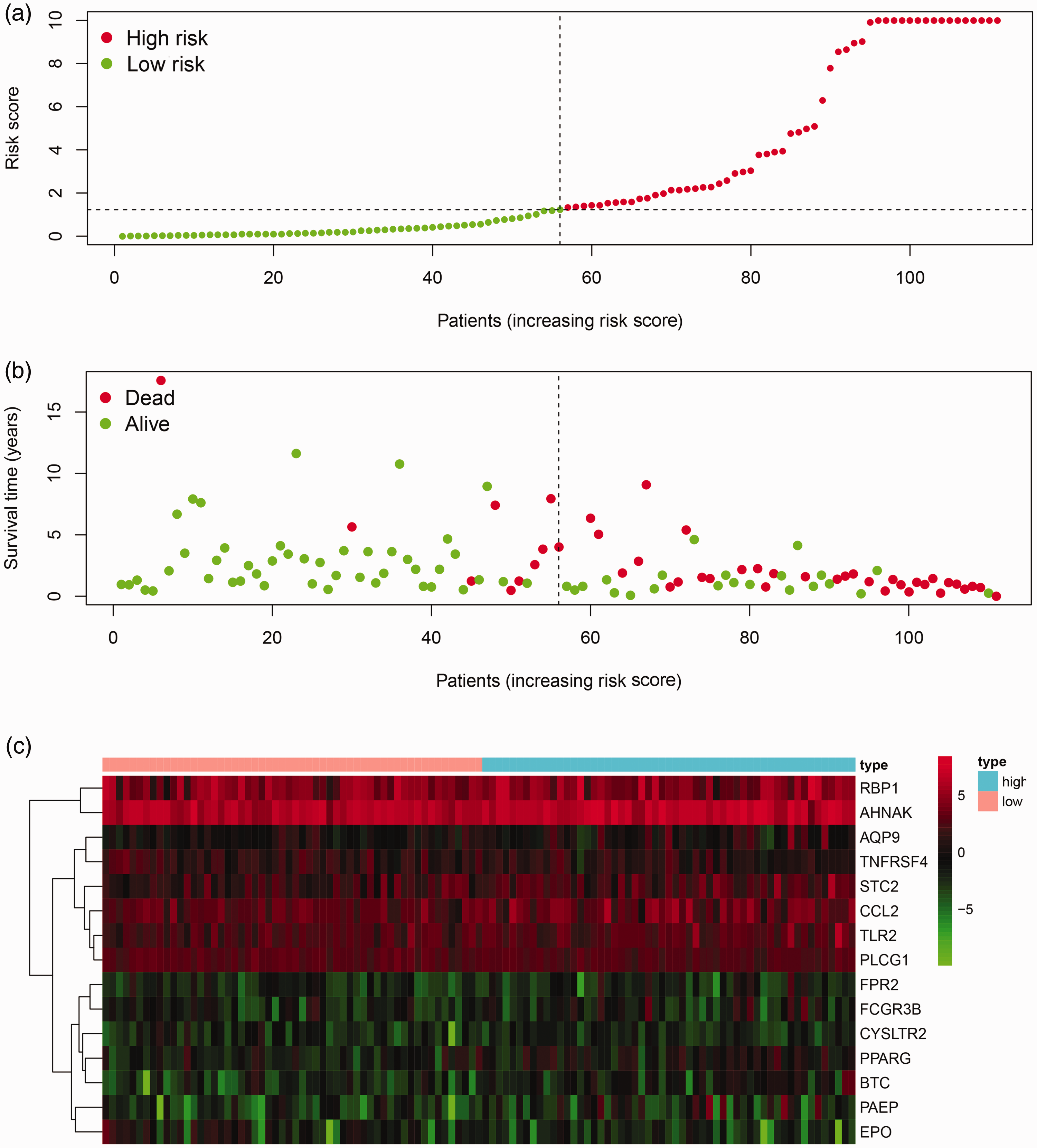
Prognostic risk score model analysis of 15 prognostic immune-related genes in patients with laryngeal cancer. (a) Risk score map: red dots represent high-risk patients; green dots represent low-risk patients. (b) Survival status distribution map: red dots represent dead patients; green dots represent alive patient. (c) Expression heat map: prognosis-associated immune genes with higher expression are shown in red, those with lower expression are shown in green, and genes that were not differentially expressed are shown in black. The bar at the top indicates patient source: blue = high-risk patient; pink = low-risk patient
Independent prognostic analysis in laryngeal cancer
Subsequently, univariate Cox regression analysis was performed to assess the relationship between independent prognostic factors and prognosis. As shown in Figure 7, risk score was significantly associated with prognosis (P < 0.001). Sex and N stage were also significantly associated with prognosis (P < 0.05). To remove any factors that might not be independent for laryngeal cancer patients, multivariate Cox regression analysis was also performed. As shown in Figure 7, risk score (P < 0.001) and sex (P < 0.05) were potential independent prognostic factors that could be applied to clinical analysis for laryngeal cancer.

Forest map of univariate and multivariate Cox regression analyses assessing independent prognostic factors age, sex, tumor grade, tumor stage, T, N, and risk score in patients with laryngeal cancer. (a) Univariate Cox regression analyses; (b) multivariate Cox regression analyses. An HR value >1 (red) indicates high risk, and an HR value <1 (green) indicates low risk
Discussion
In recent years, immune-targeting drugs are an emerging treatment method for malignancies; however, immunotherapy for laryngeal cancer is still in its infancy compared with that for other malignancies.9,10 Zeng et al. 11 identified and evaluated 23 immune-related genes in laryngeal cancer through gene co-expression networks. Zhang et al. 12 reported a novel prognostic signature containing five genes in laryngeal cancer. In this study, we constructed a regulatory network of prognosis between transcription factors and 30 prognostic immune-related genes. In the regulation network between transcription factors and immune-related genes (Figure 4), ETS1 and BATF were key transcription factors that regulated several immune-related genes. The ETS1 proto-oncogene is a member of the ETS family of eukaryotic transcription factors that play roles in a number of biological processes, including the regulation of immune-related cells, such as B and T cells.13–15 The ETS family genes and their products have been implicated in several malignant diseases and pathological genetic disorders.16,17 Studies reported that ETS1 and ETS2 have been shown to act as proto-oncogenes and promote tumor formation in nude mice.17,18 In our study, the regulation network showed that the immune-related genes AQP9, FPR2, IL13RA2, and AHNAK were positively regulated by transcription factor ETS1. Additionally, studies show that BATF family transcription factors (BATF, BATF2, and BATF3) also play important roles in the regulation of immune-related cells, such as T cells and dendritic cells.19–21 Several studies suggest that BATF2 is a novel tumor suppressor gene, which could inhibit growth of cancer cells.22–25 High BATF2 expression and overexpression of BATF2 promote growth inhibition and apoptosis in cancer cells; 22 however, low BATF2 expression is associated with significantly increased mortality in colorectal cancer, 25 hepatocellular carcinoma (HCC), 26 and oral (tongue) squamous cell carcinoma. 27 Guler et al. 24 suggest that BATF2 could be a potential therapeutic target against cancer by augmenting BATF2 in malignant cells. In our study, the regulation network showed that immune-related genes XCL2, ZAP70, TNFRSF4, LCK, TRBC1, and TRBJ2-3 were positively regulated by transcription factor BATF. Thus, these results show that ETS1 and BATF exhibit strong interactions with several immune-related genes and cancers and might be used as potential biomarkers or promising therapeutic targets for laryngeal cancer prognosis.
In this study, we explored the relationship between immune genes and prognosis in laryngeal cancer. Interestingly, we identified more upregulated (11) than downregulated (4) immune-related genes (Supplemental Table S2). One laryngeal cancer immune-related gene screened as a hub gene was RBP1 (also known as CRBP1). CRBP1 is reported to be downregulated in certain human cancer tissues, including prostate cancer, 28 breast cancer, 29 endometrial cancer, 30 and ovarian cancer, 31 and upregulated in lung adenocarcinoma 32 and laryngeal cancer.33,34 Peralta et al. 33 , 34 reported that upregulation of the CRBP1 gene and its expression correlated significantly with survival in patients with laryngeal carcinoma and that CRBP1 could have potential as a novel marker for long-term survival in laryngeal squamous cell carcinoma. In our study, as in previously published studies, RBP1 was also upregulated in laryngeal cancer. FPR2 is another laryngeal cancer immune-related genes that we identified by screening; it is overexpressed in colon cancer, 35 melanoma, 36 and ovarian cancer cells, 37 and elevated FPR2 expression is associated with poorer prognosis. Several other prognostic immune-related genes (e.g., PAEP, AHNAK, BTC, STC2) have also been reported to be associated with cancers,38–41 whereas CCL2 has been reported to be related to immunity and inflammation. 42
Finally, we established a prognostic risk assessment model based on risk scores. Both survival analysis and ROC curve analysis demonstrated that the prognostic model was beneficial for assessing poor or satisfactory survival time; detection of the expression level of genes in the model might have a robust clinical impact and help in individualized therapy and personalized laryngeal cancer management. Nevertheless, there are several limitations in our study because our conclusions are drawn only from the analysis of data in the TCGA database; further study is needed for verification.
In conclusion, we identified 15 immune-related genes by screening and established a prognostic model for laryngeal cancer. The results might indicate novel predictive biomarkers and therapeutic targets for research into the molecular mechanisms and treatment of laryngeal cancer.
Supplemental Material
sj-pdf-1-imr-10.1177_0300060520964662 - Supplemental material for Identification of prognostic immune genes in laryngeal cancer
Supplemental material, sj-pdf-1-imr-10.1177_0300060520964662 for Identification of prognostic immune genes in laryngeal cancer by Huan Xiao, Qi-sheng Su and Chao-qian Li in Journal of International Medical Research
Supplemental Material
sj-pdf-2-imr-10.1177_0300060520964662 - Supplemental material for Identification of prognostic immune genes in laryngeal cancer
Supplemental material, sj-pdf-2-imr-10.1177_0300060520964662 for Identification of prognostic immune genes in laryngeal cancer by Huan Xiao, Qi-sheng Su and Chao-qian Li in Journal of International Medical Research
Footnotes
Declaration of conflicting interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grants from the Guangxi Natural Science Foundation (No. 2020GXNSFDA238003) and the Guangxi Medical Leadership Talent Construction Foundation of China (No. 2100409).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.