
Abstract
Lipoblastomas and liposarcomas are rare causes of soft tissue masses in paediatric patients. In this retrospective clinical case series we identified 11 patients from our paediatric database (10 with a lipoblastoma and one with a liposarcoma) who had attended our hospital between 1998 and 2019. The median age of patients with lipoblastoma was 29 months. All lipoblastoma cases were managed with surgical excision and histological examination. The 18-year old patient with liposarcoma presented with a metastatic and unresectable tumour that was unresponsive to chemotherapy and radiation. Our experience demonstrates the importance of differentiating the type of soft tissue mass in children.
Introduction
Lipoblastoma is a soft tissue neoplasm, composed of immature adipocytes, thought to be derived from the imperfect embryogenesis of white fat. 1 Although the tumours are benign, risk for complications can occur because of their mass effect and when they are found in surgically challenged anatomical sites. Adipocytic tumours account for approximately 10% of soft tissue tumours in children and lipoblastomas are responsible for about 18–30% of these tumors. 2 It has been estimated that 90% of lipoblastomas occur in children under the age of three years. 2 The standard care for lipoblastoma is surgical excision with clear margins; after resection, patients should be monitored closely because of high rates of local recurrence. 2
Liposarcomas are extremely rare and account for less than 2% of all soft tissue malignancies in children with 90% of them occurring in the second decade of life. 2 They are a heterogeneous group of malignant neoplasms with variable risk of metastases and mortality depending on specific histology, underlying genetic alterations, and histological grade. 3 While liposarcomas are also lipogenic tumours, they have a distinct genetic profile that distinguishes them from lipoblastomas. 2 Wide local surgical excision is the mainstay of treatment for liposarcomas with adjuvant treatment options including chemotherapy and radiation used in cases with high-risk histology or metastasis. 2
We report a case series of 11 children with lipoblastoma or liposarcoma who attended Loma Linda University Children’s Hospital, CA, USA between 1998 and 2019.
Methods
Children up to 18 years of age who had attended our hospital between 1998 and 2019 and had a confirmed pathological diagnosis of a lipoblastoma or liposarcoma were identified and selected from our paediatric surgery divisional database. Patients’ demographics, clinical presentations, clinical courses, surgical managements, pathological diagnosis, imaging findings and outcomes were extracted from the database and anonymised. This retrospective, clinical case series was granted institutional review board approval [IRB# 5200292].
Results
Eleven patients were identified from the database, 10 with a lipoblastoma and one with a liposarcoma. The median age of patients with lipoblastoma was 29 months (range 6 months–17 years) and the male to female ratio was 9:1 (Table 1). The locations of the lipoblastomas were evenly distributed between central and peripheral sites and the peripheral tumours demonstrated a predilection for limb girdles. Four tumours were located in the abdomen, one in the flank, two in the buttock, one adjacent to the scapula, one within the axilla and one in the thigh. The sizes of the tumours ranged from 2–26 cm. Nearly all patients had a similar presentation, whereby a parent or the child had reported a lump that slowly grew over the course of six months to a year. A couple of reports described how the child’s gait had been impacted, causing the parent to seek medical attention. On physical examination, the tumours were described as firm, mobile, rubbery, fluctuant, well-demarcated and multilobulated. Prior to intervention, differential diagnoses had included cystic hygroma, lipoma and liposarcoma. Two cases had undergone a pre-operative needle biopsy due to concerns of malignancy.
Summary of reported cases.

All patients with lipoblastoma underwent surgical excision of the mass without complication. Tumours that were located deeply provided challenging operations. For example, in Case 9, after a generous midline incision, the mass was found to be intertwined with small bowel loops and was closely adhered to the mesentery; a careful and time-consuming dissection ensued. In this patient, numerous feeder vessels were found penetrating into the mass and during excision were subjected to a combination of suture ligation, clips or harmonic scalpel. To remove the base of the mass, the superior mesenteric artery and vein, the pancreas and splenic vein were identified, preserved and separated from the mass. This particular case report illustrates the importance of imaging in surgical planning which undoubtedly assists in anticipating difficulties these masses may cause. Although all patients were scheduled for a post-operative follow-up appointment, four of the ten lipoblastoma patients were lost to follow-up. Of the six remaining patients, no reports of recurrences occurred within the following 12 months.
Gross examination of the tumours showed that the cut surface was pale tan in colour (Figure 1). Histologically, the lipoblastomas demonstrated predominantly myxoid stroma containing a rich capillary network (Figures 2 and 3). The tumours showed characteristic blue grey myxoid stroma, surrounding adipocytes and occasional lipoblasts with smaller lipid vacuoles and the central lobule flanked by fibrous septae (Figure 4). An example of a high-power view shows an admixture of delicate, branching capillaries, bi-vacuolated lipoblasts, and mature adipocytes (Figure 5).

Cut surface of a gross specimen from Case 7 showing a pale tan surface.

Histologically, the lipoblastomas demonstrated predominantly myxoid stroma containing a rich capillary network, features which may lead to diagnostic confusion with myxoid liposarcoma (Case 7). Haematoxylin and Eosin (H&E) stain, 100x magnification.

Histologically, the lipoblastomas demonstrated predominantly myxoid stroma containing a rich capillary network, features which may lead to diagnostic confusion with myxoid liposarcoma (Case 7). Haematoxylin and Eosin (H&E) stain, 100x magnification.
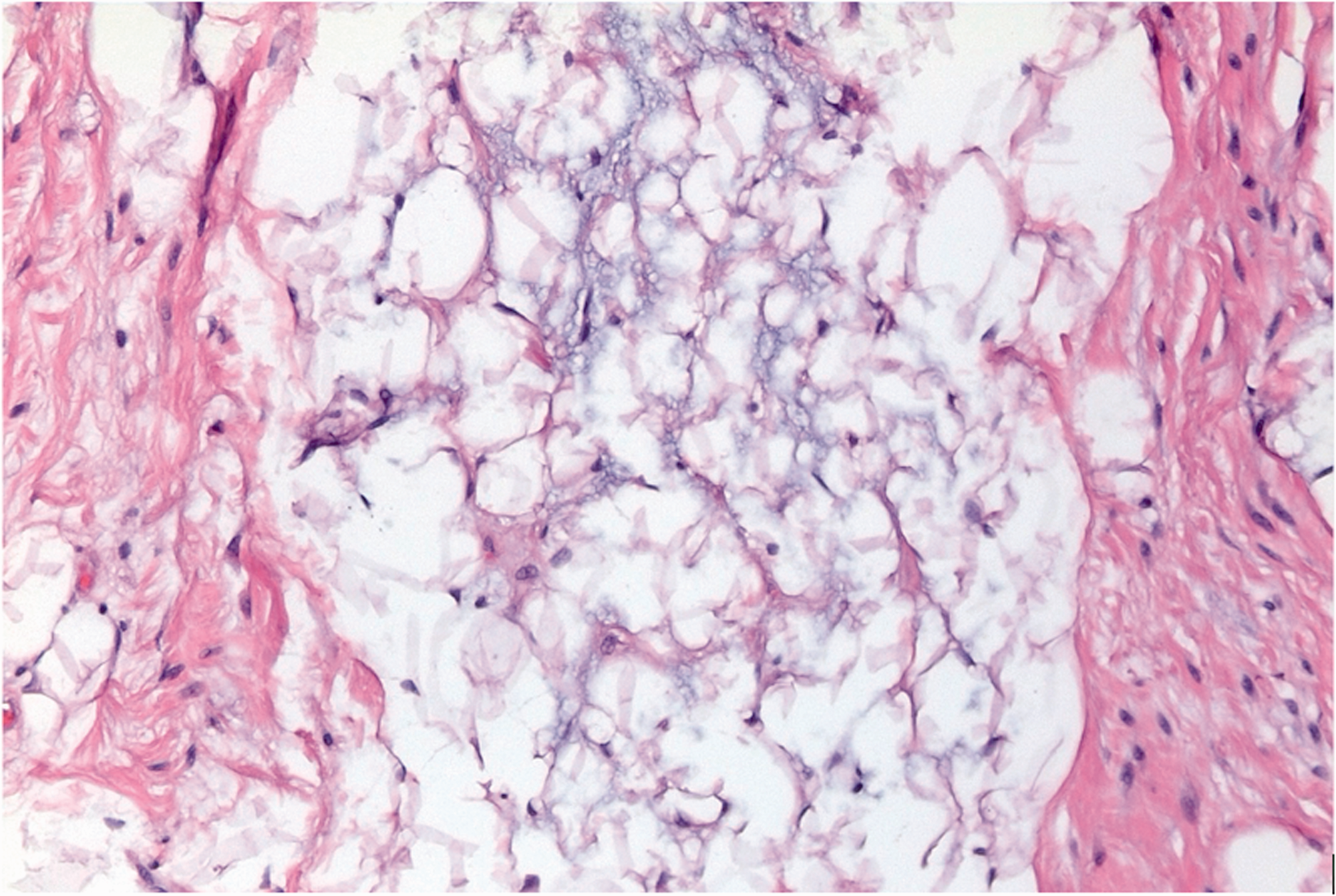
Characteristic blue grey myxoid stroma of lipoblastoma, surrounding adipocytes and occasional lipoblasts with smaller lipid vacuoles. The central lobule is flanked by fibrous septae on the sides (Case 10). Haematoxylin and Eosin (H&E) stain, 200x magnification

An example of a high-power view shows an admixture of delicate, branching capillaries, bi-vacuolated lipoblasts, and mature adipocytes (Case 9). Haematoxylin and Eosin (H&E) stain, 400x magnification.
Figure 6 shows a computed tomography (CT) scan of the abdomen and pelvis with contrast from Case 7. The yellow arrow indicates a large heterogenous and lobulated intraperitoneal mass with mixed fatty and solid components. There is no evidence of vascular invasion, visceral origination or bowel communication.

A computed tomography (CT) scan of the Abdomen and Pelvis with Contrast (Case 7). The yellow arrow is pointing to a large heterogenous and lobulated intraperitoneal mass with mixed fatty and solid components. There is no evidence of vascular invasion, visceral origination or bowel communication.
Figure 7 shows magnetic resonance imaging (MRI) of the pelvis with contrast from Case 10. A heterogenous soft tissue mass within the subcutaneous fat overlying the posterior lateral aspect of the right iliac wing was observed. There was no muscular invasion or periosteal reaction.
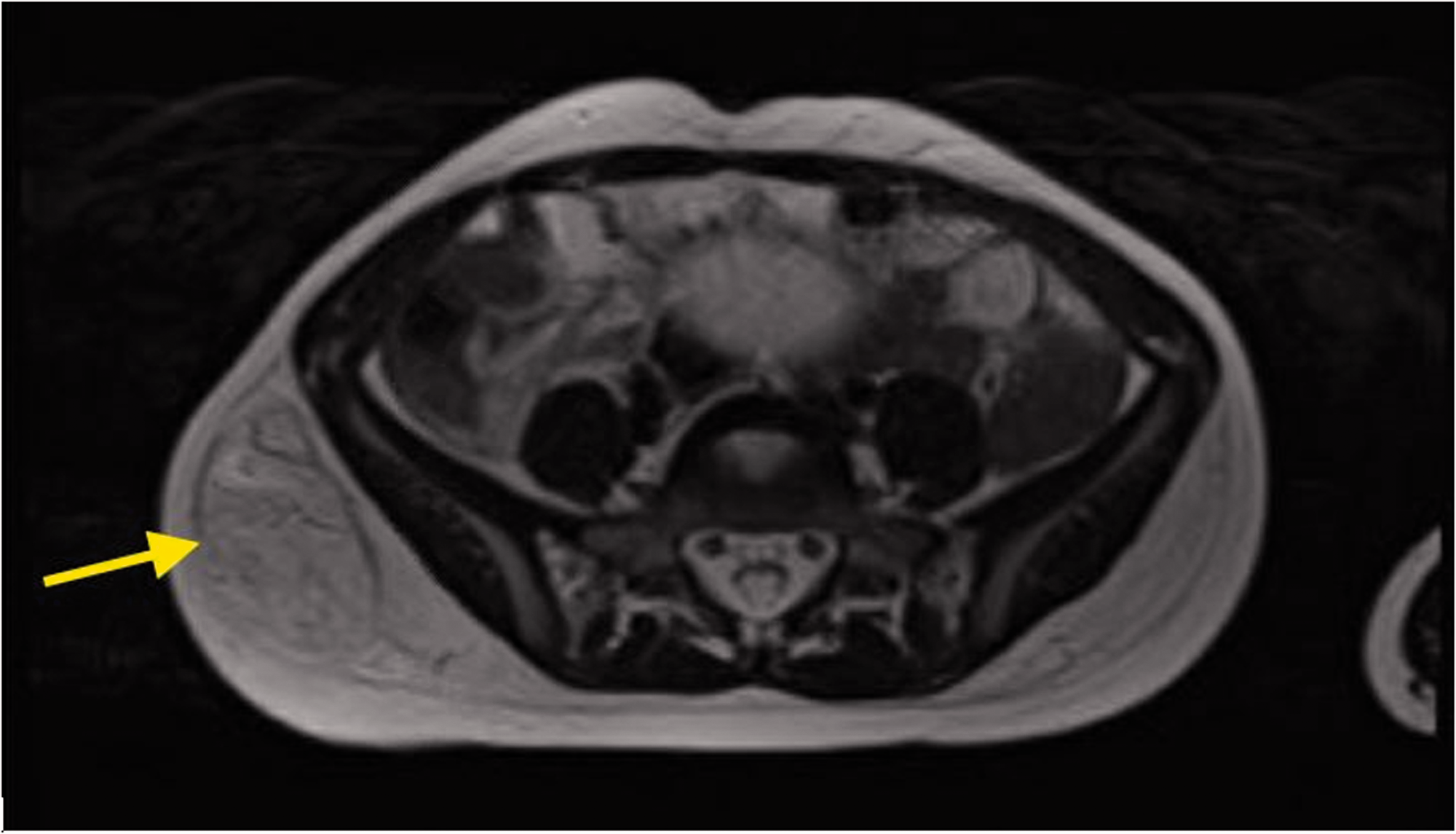
Magnetic Resonance Imaging (MRI) of the Pelvis with Contrast (Case 10). The T2 axial image shows a heterogenous soft tissue mass (yellow arrow) within the subcutaneous fat overlying the posterior lateral aspect of the right iliac wing. There is no muscular invasion or periosteal reaction.
Only one case of liposarcoma was reported. An 18-year-old man had initially presented to another hospital with right hip pain and in the subsequent work up had undergone a computed tomography (CT) scan. A large mass was detected that occupied the entire pelvic region and extended to the level of the inferior poles of both kidneys. An exploratory laparotomy and biopsy were completed and showed liposarcoma. The patient underwent chemotherapy and radiation without any significant response. He was transferred to our hospital for further management. A CT abdomen and pelvis (CTAP) demonstrated a large, multilobulated, heterogenous and necrotic mass occupying the entire pelvis and involving the pelvic floor musculature and right psoas muscle. There was a profound mass effect on the rectum, sigmoid colon and both ureters with extrinsic compression. The urinary bladder was displaced anteriorly and was closely associated with the mass. A CT of the chest showed multiple bilateral pulmonary nodules and a small low-density lesion in the left hepatic lobe. Due to the extensive nature of the patient’s disease and lack of response to previous neoadjuvant therapy, the liposarcoma was deemed unresectable and the patient was discharged to a hospice.
Discussion
Lipoblastomas are rare soft tissue tumours mostly found in young children. 2 From this retrospective case series study which spanned 21 years from 1998 and 2019, we identified 11 patients with soft tissue tumours who had attended our centre for treatment, 10 with lipoblastoma and one with liposarcoma. Most patients were male and their median age was 29 months. The patient with a liposarcoma was an 18-year-old male. All lipoblastoma cases underwent successful surgical excision of the mass with no known recurrences. The patient with liposarcoma presented with an unresectable mass that was unresponsive to chemotherapy and radiation. Our experience demonstrates the importance of differentiating the cause of a soft tissue mass in children because in adolescents and teenagers there is an increased likelihood that a lipogenic tumour may be malignant.
Our findings are consistent with those from other case series.4,5 For example, in a review of published reports from 1973–2014 that found 263 lipoblastoma cases, most patients were male with an average age of presentation of 22.6 months. 4 Another study of 12 cases reported masses in a similar location to our cohort with the exception of two detected in the groin area. 5
Lipoblastomas are of worldwide concern.5–9 They can be clinically and radiologically challenging to distinguish from other fat-containing soft tissue masses including hibernomas, lipomas and lipoma variants including spindle cell, pleomorphic lipoma and fibrolipoma, some of which are malignant. 4 In addition, they are fast growing and may result in a mass effect resulting in bothersome symptoms or cosmetic problems without adverse symptoms. 10 Moreover, complete resection of the tumours is crucial since incomplete resected tumours may regrow. 10
When a child presents with a painless enlarging mass, ultrasound is typically the first imaging modality employed. On ultrasound, lipoblastomas can appear as a well-defined hypoechoic mass with internal septations 11 or they can have mixed-echogenicity and internal cystic spaces. 12 Due to this variable appearance, ultrasound is thought to be not sensitive or specific enough to be diagnostically accurate for lipoblastoma. Although CT is often used in assessment of lipoblastomas, MRI is the preferred method of imaging due to its resolution in signal intensity but it still has some limitations. 11 On MRI, lipoblastomas are often seen as solid encapsulated and lobulated masses with a heterogenous hyperintense signal in T1- and T2-weighted sequences. 11 Due to the varying content of adipose tissue in each lipoblastoma, some may appear as lipomas with increased hypointensity on fat-suppressed images, while those with less mature adipose tissue may appear more heterogenous. 12 Myxoid liposarcoma have a small adipose component and show low signal intensity on T1-weighted images along with high signal intensity foci. 12
Although lipoblastomas can show histologic overlap with spindle cell lipoma and myxoid liposarcoma, diagnosis is typically resolved by pathological evaluation. In challenging cases, myxoid liposarcomas can be reliably diagnosed based on a recurrent t (12; 16 (q13; p11) reciprocal translocation, which results in fusion of the FUS and DDIT3 genes. 13 This fusion can be detected by either fluorescent in situ hybridization (FISH) or polymerase chain reaction (PCR) based testing. Lipoblastomas are thought to arise from an atypical embryogenesis of white fat. 1 While they are typically composed of immature adipose tissue, some specimens may show extensive maturation of the adipose tissue, from primitive stellate or spindled mesenchymal cells to multivacuolated lipoblasts, small signet ring lipoblasts or mature adipocytes. 14 Immature adipose tissues appear pale tan on gross specimens in comparison with mature adipose tissue that often appears bright yellow reflecting their high proportion of mature adipose content. Lipoblastomas typically exhibit a component of myxoid stroma, features which may lead to diagnostic confusion with myxoid liposarcoma. They also exhibit a lace-like branching capillary network low cellularity, and bland cytomorphology without atypia or readily identifiable mitotic activity. 15 While the tumour cells are diffusely positive for S100 and CD34 immunohistochemical stains, these are not typically necessary to make a diagnosis and are not useful in distinguishing them from histological mimics. 16 Cytogenetically, most lipoblastomas have been shown to demonstrate a rearrangement on chromosome 8q12 within the PLAG 1 gene, or sometimes in the HMGA2 gene, resulting in gene upregulation, which can most reliably be detected by RNA sequencing. However, PLAG1 FISH or PLAG1 immunohistochemistry is a more practical diagnostic modality than PCR based testing.15,17
The treatment of lipoblastoma is surgical resection with free margins to decrease risk of recurrence. 18 Recommendations suggest that patients should be followed-up after intervention for at least five years with a suggestion of up to ten years because of the high rates of recurrence that are often delayed. 18 To date, there have been no cases of mortality associated with lipoblastoma, but morbidity can increase because of the mass effect caused by the rapid growth of the tumour. 4
As observed in our case series, liposarcoma is even rarer than lipoblastoma in the paediatric population. Liposarcomas are significantly more prevalent in older children and adults than young children or infants. 2 Indeed, fewer than 3% of all liposarcoma cases have been reported in children. 19 It is important to distinguish between the two types of soft tissue tumours because of the malignant nature of liposarcomas. In one review of liposarcoma outcomes, prognosis was dependent on the grade and type of tumour; the high-grade pleomorphic sub-type had a median survival of 1.9 years whereas low-grade myxoid and well-differentiated subtypes had median survival times of 7.3 and 5.4 years, respectivley. 19 In addition, eight reported deaths were associated with anatomically centralized lesions, similar in location to the case study in our report. 19
Histologically, liposarcomas often have irregular or incomplete fibrous septa, a peripheral concentration of mature adipose cells within lobules, higher cellularity and abnormal mitoses. 14 There are five histological subtypes of liposarcoma that can be grouped into three main categories: well-differentiated/dedifferentiated; myxoid/round cell; pleomorphic.3,19 Differentiating between the tumour subtypes can assist with the treatment plan. Moreover, differentiating between malignant myxoid liposarcoma and benign lipoblastoma is extremely important. In histologically challenging cases, genetic testing may be helpful. For example, the vast majority of myxoid liposarcomas have a cytogenetic rearrangement at chromosome 12q13 (DDIT3) with either 16p11 or 22p11, which does not occur in lipoblastomas. 14
The treatment for liposarcoma is also surgical excision, but in cases where surgical management is insufficient or too great a risk for morbidity, the different subtypes have varied responses to chemotherapy or radiation. 3 For example, well-differentiated/dedifferentiated liposarcomas have been shown to have low chemosensitivity and moderate radiosensitivity. By contrast, myxoid/round cell liposarcomas have high sensitivity to both chemotherapy and radiation, and pleomorphic liposarcoma has high chemosensitivity and moderate radiosensitivity. 3 However, unlike adults with liposarcoma, optimal radiation dosages and the role of chemotherapy has not been well-established for children. 19 Targeted therapies such as CDK4 inhibitors and FUS-DDIT3 binding inhibitors (e.g., trabectedin) are two treatment options that are being investigated for the treatment liposarcoma. 3
In summary, in this retrospective review of patient records, we identified ten cases of lipoblastoma and one of liposarcoma from our paediatric data collected between 1998 and 2019. All lipoblastoma cases underwent successful surgical excision of the mass with no known recurrences. The one patient with liposarcoma presented with a metastatic and unresectable tumour that was unresponsive to chemotherapy and radiation. Our experience demonstrates the importance of differentiating the type of soft tissue mass in children because in adolescents and teenagers there is a high risk of a lipogenic tumour being malignant.