
Abstract
Objectives
We investigated the endoplasmic reticulum (ER) stress markers C/EBP homologous protein (CHOP) and glucose-regulated protein (GRP) 78, as well as the inflammatory factors nuclear factor (NF)-κB and IκBα, to assess how social defeat stress induces myocardial injury. Furthermore, we evaluated the protective effects of the ER stress inhibitor 4-phenylbutyric acid (PBA) on myocardial injury in mice.
Methods
Adult mice were divided into control, control + PBA, social defeat, and social defeat + PBA groups. The social defeat and social defeat + PBA groups were exposed to social defeat stress for 10 days. Cardiac tissues from all groups were analyzed after social defeat stress. H9C2 cells were used to detect the role of the ER stress agonist thapsigargin on expression of ER stress and inflammatory markers.
Results
Social defeat stress promoted apoptosis of cardiomyocytes, increased CHOP, NF-κB and, phospho-NF-κB protein expression, and decreased GRP78 and IκBα protein expression. Moreover, PBA significantly reversed these changes and attenuated thapsigargin-induced increased expression of CHOP and phospho-NF-κB, and decreased IκBα expression in H9C2 cells.
Conclusions
Social defeat stress initiates ER stress, promotes expression of inflammatory factors, and induces myocardial injury. Inhibiting ER stress could protect the myocardium from social defeat stress-induced myocardial injury.
Keywords
Introduction
Social stress is a causal factor for depression and cardiovascular disease, and the etiologies of these two disorders are related.1–3 Depression significantly increases mortality from cardiovascular disease, while cardiovascular disease exacerbates depression. Animal models have shown that stress elevates blood pressure and plasma catecholamine levels.4–6 Additionally, increased heart rate by social stress is strongly associated with ventricular and supraventricular arrhythmias. 7 We have previously shown a strong correlation between social defeat stress, cognitive dysfunction, and depression.8,9 Other studies have identified increased inflammatory factors in depressed individuals.10–13
The endoplasmic reticulum (ER) is the cellular organelle responsible for protein translation, folding, and transport, and is also involved in many cellular signaling pathways. ER stress is induced by multiple factors including high glucose, energy deprivation, oxidative stress, hypoxia, and calcium overload. Several studies have demonstrated that ER stress promotes diabetes, neurodegenerative diseases, viral infections, cancer, inflammatory diseases, and cardiovascular disease.14–16 Chen et al. 17 showed that an increase in glucose-regulated protein (GRP) 78, GRP94, and calreticulin in the temporal cortex were positively correlated with the risk of suicide in patients with depression. These findings suggested a potential role for ER stress-related proteins in depression. Depression is a major risk factor in the etiology of cardiovascular disease. Our previous study showed that social defeat stress altered expression of ER-related proteins, including the anti-apoptotic protein GRP78 and the apoptotic transcription factor C/EBP homologous protein (CHOP), in the brains of adolescent mice. 9 ER stress-induced inflammation and cell apoptosis play important roles in regulating chronic stress responses and changing the structure and morphology of the brain.8,9,18
In addition to a sedentary lifestyle and traditional cardiovascular risk factors, such as smoking, hypertension, and hyperlipidemia, chronic inflammation induced by social stress plays an important role in increasing the risk of coronary heart disease.19,20 Arati et al. 21 showed that acute stress stimulation altered mouse behavior, increased body temperature, and induced inflammation in the myocardium. However, whether inflammatory factors contribute to social defeat stress-induced myocardial injury is unclear.
To investigate the molecular mechanisms by which social defeat stress causes myocardial injury, we analyzed two major targets of the important inflammatory factor nuclear factor κB (NF-κB) and the ER stress regulator CHOP. Expression of these two proteins indicates the level of ER stress and the effect of ER stress on inflammatory responses. We investigated the effect of social defeat stress on myocardial injury by evaluating apoptosis of cardiomyocytes and expression of myocardial inflammatory factors. We also analyzed the effect of the ER stress inhibitor 4-phenylbutyric acid (PBA) on myocardial injury.
Methods
Materials
PBA and thapsigargin (TG) were purchased from Sigma (Sigma-Aldrich, St. Louis, MO, USA). All biochemical reagents were purchased from Sigma-Aldrich and antibodies were purchased from cell signaling (Cell Signaling Technology, Inc., Danvers, MA, USA) unless otherwise indicated.
Animals and treatment protocols
The animals used in this study included 7-week-old inbred male C57BL/6J mice (total of 24, n = 8 in each group) and 14-week-old Institute of Cancer Research (ICR)-occluded mice. All mice were housed in groups (eight mice per cage) in a temperature-controlled room at 22°C ± 2°C, with 40% to 60% humidity, under a 12-hour light/dark cycle (lights on from 07:00–19:00 hours) with food and water ad libitum before the social defeat experiment. The mice were randomly divided into the following four groups: 1) control; 2) control + PBA; 3) social defeat; and 4) social defeat + PBA. The control + PBA and social defeat + PBA groups were treated with 100 mg/kg PBA via intraperitoneal injection according to a previous study.22,23 The control and social defeat groups were treated with an equal amount of vehicle based on body weight. All mice were treated once a day for 2 days before the start of the social defeat stress. All experiments were conducted in accordance with the Guidelines for Animal Experiments of The Second Affiliated Hospital of Xinxiang Medical University.
Cell culture and treatment
H9C2 cells were purchased from Clonetics (San Diego, CA, USA) and maintained in Dulbecco’s modified Eagle’s medium (Invitrogen Life Technologies, Grand Island, NY, USA), supplemented with 10% fetal bovine serum (Invitrogen Life Technologies), 1% penicillin-streptomycin, and 1% L-glutamine. H9C2 cells from the fourth to the ninth passages were used throughout the study. PBA and TG were prepared in dimethyl sulfoxide and immediately diluted with the culture medium before the experiment.
H9C2 cells were grown to 70% to 80% confluence in OPTI MEM medium (Invitrogen Life Technologies) and were then incubated with TG (500 nM) for 24 hours in the presence or absence of PBA (500 nM). 24 When PBA was included, it was added 1 hour before TG incubation. After incubation, the cells were collected to assess protein expression by western blotting.
Social defeat stress
To create a social stress model, reliably aggressive ICR mice (three consecutive attacks within 30 s) were selected as the aggressor mice. The social defeat group (7-week old C57BL/6J mice, n = 8) was physically exposed to a different aggressor for 10 minutes every day for 10 days. After 10 minutes, the defeated mice were subjected to continuous psychological stress from sensory interaction (smell and sight of the aggressor) with the aggressor for the remainder of the 24-hour period through a clear perforated divider in a shared home cage. All social defeat mice were rotated on a daily basis to ensure that they were defeated by a different aggressor mouse every day during the 10-day period. The control mice were housed with a clear perforated divider in a shared home cage, but with members of the same strain that were changed daily. All control mice were rotated on a daily basis and physical contact with their cage mate was avoided.
Preparation of heart tissue
After the social defeat protocol was concluded (day 11) (Figure 1), the mice were euthanized by decapitation under ether anesthesia. The heart was rapidly removed and washed with ice cold saline. The left ventricles were used for histological measurements and the right ventricles were used for apoptosis and biochemical analysis. The remaining heart tissue was preserved at −80°C for western blotting.

Experimental schedule. The mice were randomly divided into the control, control + PBA, social defeat, and social defeat + PBA groups. The control + PBA and social defeat + PBA groups were treated with 100 mg/kg PBA and the control group and the social defeat groups received an equal amount of vehicle on the basis of body weight. All mice were treated via intraperitoneal injection once a day for 2 days before the start of social defeat stress.
Western blotting
Tissue samples were homogenized in 20 mM ice-cold Tris-HCl (pH 7.4) containing 1% protease and phosphatase inhibitors. The homogenates were centrifuged for 15 minutes at 18,000 × g at 4°C and the resulting supernatant fractions were used for western blotting. The protein samples (20 μg/lane) were separated and transferred to hydrophobic polyvinylidene difluoride membranes. The membranes were blocked and incubated overnight at 4°C with monoclonal rabbit anti-GRP78, anti-phospho-NF-κB antibody (1:1,000), monoclonal mouse anti-CHOP, anti-NF-κB antibody (1:1,000), or monoclonal rabbit anti-IκBα (NF-κ inhibitor, alpha) antibody (1:1000; Abcam, Cambridge, UK) in 5% non-fat milk. The next day, the membranes were washed with phosphate-buffered saline + Tween, and the primary antibody was detected using a horseradish peroxidase-conjugated goat anti-rabbit IgG antibody (1:20,000; Vector, Burlingame, CA, USA) or horseradish peroxidase-conjugated horse anti-mouse IgG antibody (1:5000) in phosphate-buffered saline for 60 minutes at 25°C. The blots were developed using an enhanced chemiluminescence reagent (GE Healthcare, Inc., Piscataway, NJ, USA). The blots were then visualized with a LAS-3000 Plus lumino-imaging analyzer (Fuji Photo Film Company, Kanagawa, Japan) and quantified using Multi Gauge software v3.0 (Fujifilm, Tokyo, Japan).
Analysis of apoptosis by flow cytometry
Myocardial tissue was placed in saline, with a saline to tissue mass ratio of 1:9. The tissue was ground and filtered through a 200-mesh strainer. Cells were re-suspended at a final concentration of 2 × 107 cells/mL and the cell suspension was incubated with an annexin-V/propidium iodide (PI) staining kit (BD Biosciences, Miami, FL, USA) in the dark for 15 minutes at room temperature. The samples were then analyzed by flow cytometry (Cytomics FC 500 Beckman Coulter; Becton-Dickinson, San Jose, CA, USA).
Biochemical measurements
Heart tissues were crushed and dissolved in 0.9% physiological saline, fully ground to generate tissue homogenates, centrifuged at 4500 ×g for 10 minutes, and used to determine isoenzyme activity. The activity of creatine kinase (CK), lactate dehydrogenase (LDH), and aspartate transaminase (AST) was measured spectrophotometrically (Beckman DU640; Hercules, CA, USA) in a blinded manner according to the manufacturer’s instructions (Jiancheng Bioengineering Institute, Nanjing, China).
Histological studies
After social defeat stress, the heart was removed and washed with ice cold physiological saline. The heart tissue was fixed in 4% paraformaldehyde, dehydrated in graded ethanol, and embedded in paraffin. The paraffin-embedded tissues were cut into serial sections (thickness, 5 μm). Hematoxylin and eosin staining was then used for histomorphometric evaluation.
Heart rate and blood pressure
Heart rate and blood pressure were measured by a noninvasive computerized tail cuff system (BP-2000, VisiTech Systems, Apex, NC, USA), which was adapted for awake mice, by following the manufacturer’s instructions. 25 Measurements were conducted in a quiet area with a suitable temperature and humidity, where mice were accommodated for at least 1 hour before experiments started. Following 10 preliminary measurements in a pre-warmed tail-cuff (36°C) device to accustom mice to the procedure, 10 actual measurement cycles were collected and averaged for each individual mouse.
Statistical analysis
The results are presented as mean ± standard error of the mean. Differences among the four groups were analyzed using one-way ANOVA or two-way repeated-measures ANOVA. Pearson’s correlations were used to assess relationships between various parameters in all mice. Differences were considered statistically significant at p < 0.05. Data were analyzed with SPSS version 12.0 (SPSS, Inc., Chicago, IL, USA).
Results
Social defeat stress induces subordination of mice
During the social defeat procedure, all ICR mice attacked the intruder C57BL/6J mice (n = 8). C57BL/6J mice were all defeated and showed signs of subordination, such as sideways or upright submissive postures, freezing, fleeing, lying on the back, or withdrawal (Figure 1).
Social defeat stress induces apoptosis
Annexin V/PI staining is frequently used to identify the proportion of cells within a population that are alive, early apoptotic, late apoptotic, and dead through either apoptosis or necrosis. Therefore, we used annexin V/PI flow cytometry to assess the effect of social defeat stress on apoptosis of cardiomyocytes. The social defeat group showed a significantly higher number of cardiomyocytes in the early and late stages of apoptosis compared with the control and control + PBA groups (both p < 0.05). Additionally, the number of living cells was significantly lower in the social defeat group (Figure 2a) compared with the control group (p < 0.05), which suggested that social defeat induced apoptosis of cardiomyocytes. The social defeat + PBA group showed a significantly higher number of living cells compared with the social defeat group (Figure 2b) (p < 0.05). The social defeat + PBA group also showed a significantly lower percentage of cardiomyocyte apoptosis at the early (Figure 2c) and late (Figure 2d) stages (both p < 0.05).

The effects of social defeat stress on apoptosis of cardiomyocytes and survival. Cell apoptosis was measured by flow cytometry following annexin-V and propidium iodide (PI) double staining in cardiomyocytes (a). The lines divide each plot into the following four quadrants: lower left quadrant, living cells; lower right quadrant, early apoptotic cells; upper left quadrant, necrotic cells; and upper right quadrant, late apoptotic cells. Bar graphs summarize the mean percentage of (b) living cardiomyocytes, (c) early apoptotic cardiomyocytes, and (d) middle and late apoptotic cardiomyocytes (n = 8 per group) *p < 0.05 vs. the control and control + PBA groups; #p < 0.05 vs. the social defeat group. PBA, 4-phenylbutyric acid; FITC, fluorescein isothiocyanate.
Social defeat stress induces myocardial injury
Histopathological analysis of heart tissue from the control group showed complete structural organization of cardiac tissue, with myocardial cells that were neatly and densely arranged. In contrast, hearts from the social defeat group had disordered myocardial fibers with extensive edema, congestion, and inflammatory cell infiltration. This damage was reduced by administration of PBA (Figure 3a). CK, LDH, and AST levels are common biomarkers for diagnosing myocardial infarction and myocardial damage. 26 Therefore, we compared activity of these markers in cardiac tissue of mice in the control and social defeat groups. Enzyme activity of CK (Figure 3b), LDH (Figure 3c), and AST (Figure 3d) was significantly elevated in heart tissue from mice in the social defeat group compared with that in the control and control + PBA groups (all p < 0.05). Administration of PBA significantly attenuated CK, LDH, and AST activity compared with the social defeat group (all p < 0.05). To evaluate changes in cardiac blood volume, we measured heart rate and blood pressure. Social defeat stress significantly increased the level of heart rate (Figure 3e) and blood pressure (all p < 0.05 compared with the control and control + PBA groups) (Figure 3f), and these were alleviated by treatment of PBA (both p <0.05 compared with the social defeat group).

Effects of social defeat stress on myocardial injury. Hematoxylin and eosin staining (×200) (a) of myocardial tissue shows muscle fibers with inflammatory cell infiltration and marked edema in the social defeat group, which was alleviated by treatment of PBA. The control mouse heart shows normal structure of myocytes. Increased activity of CK (b), LDH (c), and AST (d) was observed in the social defeat group, which was attenuated by administration of PBA. Similarly, administration of PBA attenuated increased heart rate (e) and blood pressure (f), which were caused by social defeat stress (n = 8 per group) *p < 0.05 vs. the control and control + PBA groups; #p < 0.05 vs. the social defeat group. PBA, 4-phenylbutyric acid; CK, creatine kinase; LDH, lactate dehydrogenase; AST, aspartate transaminase.
Social defeat stress induces ER stress
To determine the role of ER stress in the effects of social defeat stress on myocardial injury, we measured the levels of CHOP and GRP78, which are major cellular markers of ER stress, in cardiac tissue from the four groups of mice. The pro-apoptotic transcription factor CHOP is activated by ER stress, whereas GRP78 reduces ER stress levels and apoptosis by enhancing cellular folding. Western blotting showed significantly lower GRP78 protein expression in the social defeat group compared with the control and control + PBA groups (both p < 0.05), but downregulation of GRP78 was alleviated by PBA treatment in heart tissue (p < 0.05 compared with the social defeat group) (Figure 4a, b). However, CHOP protein expression was significantly higher in the social defeat group compared with the control and control + PBA groups (both p < 0.05). Additionally, CHOP protein expression was significantly lower in the social defeat + PBA group compared with the social defeat group (p < 0.05) (Figure 4a, c). These findings suggested that social defeat stress induced ER stress in cardiac tissue of mice.

Effects of social defeat stress on endoplasmic reticulum stress marker levels in the mouse heart. Prolonged social defeat stress induced decreased GRP78 (a, b) and increased CHOP (a, c) protein expression and these were effects were attenuated by PBA administration. The ratio of CHOP/β-actin and GRP78/β-actin over the control value was used to represent the level of protein expression (n = 8 per group) *p < 0.05 vs. the control and control + PBA groups; #p < 0.05 vs. the social defeat group. PBA, 4-phenylbutyric acid; GRP78, glucose-regulated protein 78; CHOP, C/EBP homologous protein.
Social defeat stress increases NF-κB expression and reduces IκBα expression
NF-κB is a DNA-binding protein that modulates ER stress and inflammatory responses, which both play an important role in cardiovascular disease. Western blotting showed that NF-κB and phospho (p)-NF-κB protein expression was significantly different among the four groups (Figure 5a). NF-κB (Figure 5b) and p-NF-κB (Figure 5c) protein expression was significantly higher in the social defeat group compared with control and control + PBA groups (all p < 0.05). However, treatment of PBA attenuated the social defeat stress-induced increase in NF-κB and p-NF-κB expression compared with the social defeat group (both p < 0.05). This finding suggested that ER stress played an important role in the observed effects. In contrast, IκBα (nuclear transcription factor that inhibits transcription of NF-κB-dependent pro-inflammatory and apoptotic genes) protein expression was significantly lower in the social defeat group compared with the control and control + PBA groups (both p < 0.05). This effect in the social defeat group was alleviated by PBA treatment (p < 0.05) (Figure 5d).
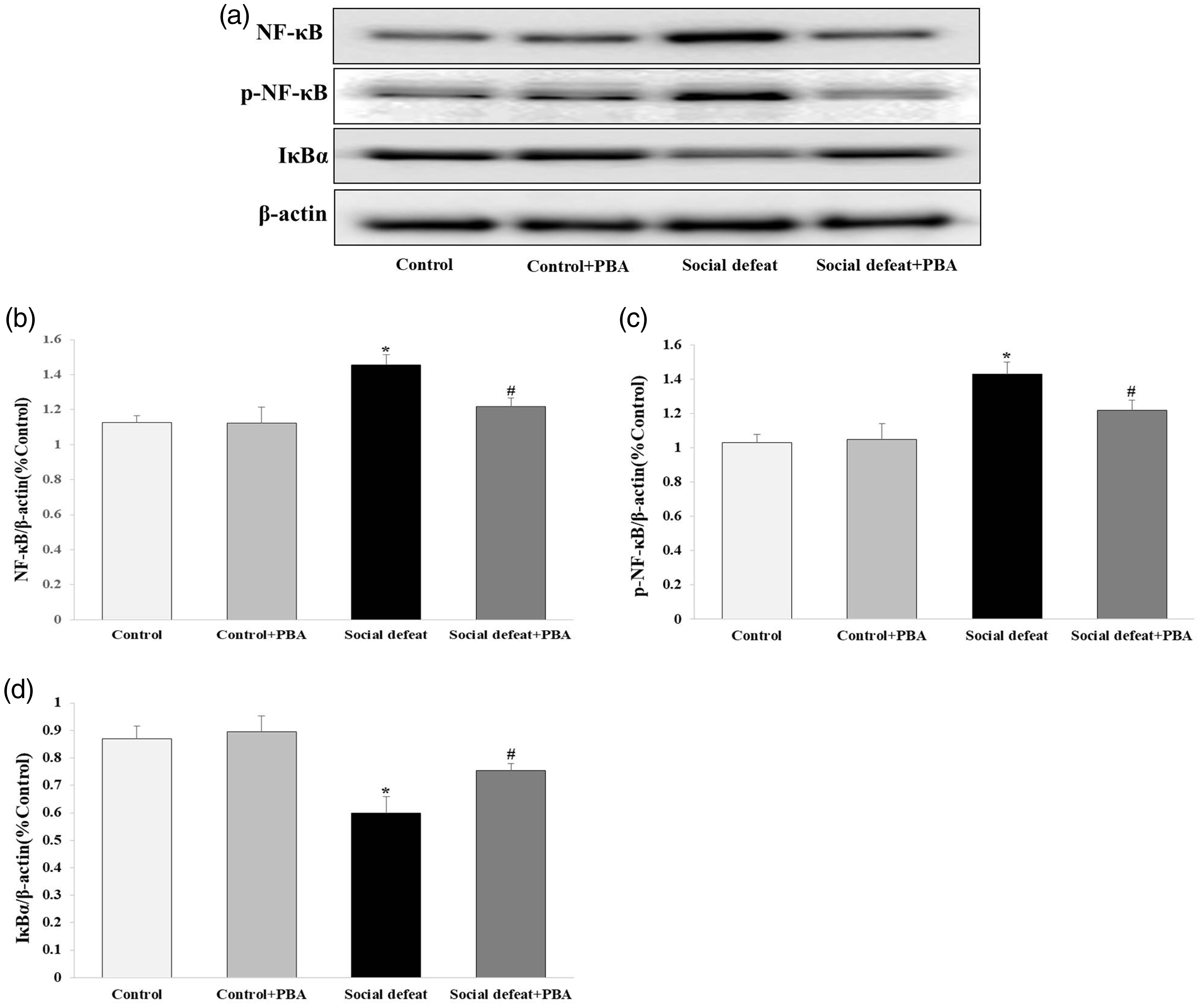
Effects of social defeat stress on inflammatory factors in the mouse heart. NF-κB (a, b) and p-NF-κB (a, c) protein expression was increased and IκBα (a, d) protein expression was decreased by social defeat stress in the mouse heart. PBA administration attenuated these changes in expression. The ratio of NF-κB/β-actin, p-NF-κB/β-actin, and IκBα/β-actin over the control value was used to represent the level of protein expression (n = 8 per group) *p < 0.05 vs. the control and control + PBA groups; #p < 0.05 vs. the social defeat group. PBA, 4-phenylbutyric acid; NF-κB, nuclear factor-κB; p-NF-κB, phospho-nuclear factor-κB.
Thapsigargin induces ER stress and NF-κB expression in H9C2 cells
To confirm the role of ER stress on inflammatory factor expression, we added TG to the culture medium to induce ER stress. Administration of TG dramatically increased CHOP protein expression (Figure 6a, b) and also increased the expression of NF-κB (Figure 6a, c) and p-NF-κB (Figure 6a, d) and decreased expression of IκBα in H9C2 cells (all p < 0.05 vs the control and control + PBA groups) (Figure 6a, e). However, treatment of PBA alone did not affect the expression of CHOP, NF-κB, p-NF-κB and IκBα. Pretreatment of PBA attenuated the TG-induced increased expression of CHOP, NF-κB, and p-NF-κB, and decreased expression of IκBα in H9C2 cells (p < 0.05 vs the social defeat group [TG]). These results showed that ER stress was induced by activating CHOP and NF-κB and this was alleviated by PBA. The results corresponded with the in vivo study.

Effects of TG on inflammatory factors in H9C2 cells. The endoplasmic reticulum stress agonist TG dramatically increased protein expression of the endoplasmic reticulum stress marker CHOP (a, b). TG also increased protein expression of the inflammatory factors NF-κB (a, c) and p-NF-κB (a, d), and decreased IκBα (a, e) protein expression. These changes in expression were attenuated by pretreatment of PBA. The ratio of CHOP/β-actin, NF-κB/β-actin, p-NF-κB/β-actin, and IκBα/β-actin over the control value was used to represent the level of protein expression (n = 8 per group) *p < 0.05 vs. the control and control + PBA groups; #p < 0.05 vs. the social defeat group. PBA, 4-phenylbutyric acid; NF-κB, nuclear factor-κB; p-NF-κB, phospho-nuclear factor-κB; CHOP, C/EBP homologous protein; TG, thapsigargin.
Discussion
Previous studies have shown that social defeat stress can induce a variety of cardiac events, such as stimulating myocardial ischemia, impairing cardiomyocyte activity, and altering myocardial structure and function. 27 Consistent with these findings, a recent study demonstrated that stress affects emotional status and leads to cellular injury in various tissues, including cardiomyocytes, which consequently increases the incidence of cardiovascular disease and mortality. 28 Similarly, several studies have indicated that ER stress-related proteins are involved in depression and bipolar disorder. 8 A previous study also showed that GRP78 and CHOP expression was significantly and positively correlated with social defeat stress-induced cognitive dysfunction. 9 However, the relationship between social defeat stress-induced myocardial injury and the expression of ER stress-related proteins is unclear.
In this study, we investigated the effect of ER stress-related proteins on cardiomyocytes using a social defeat stress mouse model and intraperitoneal injection with the ER stress inhibitor PBA. The social defeat group showed a higher number of apoptotic cells and ischemia/reperfusion injury compared with the control group. In contrast, the social defeat + PBA group showed more viable cardiomyocytes and a lower percentage of apoptotic cells compared with the social defeat group. There was no difference in the percentage of apoptotic cells between the social defeat + PBA group and the control group. The tissue specificity of various cardiac enzymes enables them to be ideal markers of tissue damage. Therefore, myocardial damage can be assessed by measuring the activity of cardiac marker enzymes. 29 In the present study, CK, LDH, and AST activity was significantly higher in the social defeat group compared with the control group, and this increase was alleviated by treatment with PBA. Histopathological studies were performed to confirm the biochemical findings. Together, these results suggested that ER stress played a major role in stress-induced myocardial injury. Therefore, inhibiting ER stress could be a potential therapeutic target in patients with a variety of cardiovascular diseases.
GRP78 and CHOP are major factors involved in ER stress. 9 Wang et al. 30 showed that GRP78 was essential in development of the heart and in maintaining cardiac contractility. In our study, the social defeat group showed lower GRP78 protein expression, but elevated CHOP protein expression, and greater myocardial injury compared with the control group. These results suggested that ER stress played an essential role in social defeat-induced myocardial injury. Treatment with the ER-stress inhibitor PBA alleviated the adverse changes in GRP78 and CHOP expression, and reduced the subsequent social defeat stress-induced myocardial damage. Moreover, inhibition of ER stress protected cardiomyocytes from the apoptosis induced by social defeat stress injury.
Because the incidence and mortality of cardiovascular disease are consistently increasing every year, they have received increasing attention. Recent studies have shown that social stress plays an important role in the onset and progression of hypertension, atherosclerosis, and arrhythmia.28,30 Cellular inflammatory responses in specialized cells and tissues activated by ER stress might participate in the pathogenesis of many diseases, such as obesity, diabetes, and atherosclerosis, which are the leading causes of cardiovascular disease.31,32 In response to ER stress, NF-κB activation can be directly promoted through the protein kinase R-like endoplasmic reticulum kinase-CHOP pathway. 33 Recent studies have shown that the ER stress-induced CHOP pathway exacerbates myocardial ischemia/reperfusion injury by inducing myocardial inflammation, which is mainly mediated by inducing NF-κB activation. Moreover the ER stress inhibitor PBA reduces hypoxia/reoxygenation-stimulated cardiomyocyte injury by reversing decreased IκBα expression in cardiomyocytes subjected to hypoxia/reoxygenation treatment.34,35
In the current study, social defeat stress increased CHOP protein expression. Social defeat stress also increased NF-κB and p-NF-κB protein expression and decreased IκBα protein expression in the mouse heart. These changes were attenuated by PBA. However treatment of PBA alone did not affect CHOP, NF-κB, p-NF-κB and IκBα protein expression. Pretreatment of PBA attenuated the TG-induced increased expression of CHOP, NF-κB, and p-NF-κB, and decreased expression of IκBα in H9C2 cells. These findings suggest that inflammatory factors are involved in social defeat stress-induced myocardial injury, which is mediated by ER stress. These results are consistent with a study by Chen et al. 36 who showed that alleviating ER stress-induced inflammation was essential for preventing cardiovascular disease. These authors also showed that increased expression and activation of NF-κB play an important role in ER stress-induced inflammation.
Our study provides important insight into the relationship between ER-associated proteins and the inflammatory consequences of cardiovascular disease induced by social defeat stress. However, there are some limitations of this study. First, the effects of exposure to an aggressor mouse in the resident’s cage were not evaluated independently. However, a similar model was used previously in which there was no effect on social interaction in mice that were exposed to an aggressive resident in the absence of social defeat 37 Second, the current study showed a relationship between myocardial injury and social defeat stress. However, whether other types of stress, such as the forced swimming test or sleep deprivation, can induce myocardial injury remains unclear. Third, we showed that starting PBA administration 2 days before social defeat stress had a myocardial protective role. However, whether PBA administration after myocardial injury can reverse the damage induced by social defeat stress also needs to be determined.
In conclusion, social defeat stress induces ER stress and promotes apoptosis of cardiomyocytes. Activation of inflammatory factors plays a major role in social defeat stress-induced myocardial injury. Therefore, inhibiting ER stress and downregulating inflammatory factors may substantially attenuate myocardial injury.
Footnotes
Authors’ contributions
Xiaolei Gao mainly contributed to design of the study, performed the experiment, and drafted the manuscript. All authors made substantial contributions to the following tasks: (1) acquisition and analysis of experimental data, (2) critical and intellectual comments about understanding the results, and (3) revising the article for submission.
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Funding
This research was partly supported by the Fund of Biomedical Research Institute, Jeonbuk National University Hospital.