
Abstract
Blast crisis develops in a minority of patients with chronic myeloid leukemia even in the era of tyrosine kinase inhibitor (TKI) therapy. Reports suggest that we know little about the mechanism of BCR-ABL and AML1-ETO co-expression in blast crisis of chronic myeloid leukemia, and that other chromosomal abnormalities also coexist. Here, we document an unusual and interesting case of a 51-year-old female diagnosed in the chronic phase of chronic myeloid leukemia. After undergoing TKI treatment for 3 months, her bone marrow aspirates in the chronic phase had transformed to blast crisis. Molecular genetic testing indicated she was positive for p210 form of BCR-ABL (copy number decreased from 108.91% to 56.96%) and AML1-ETO fusion (copy number, 5.65%) genes and had additional chromosomal abnormalities of t(8; 21)(q22; q22)/t(9; 22)(q34; q11), t(2; 5)(p24; q13) and an additional +8 chromosome.
Introduction
Chronic myeloid leukemia (CML) is characterized by the presence of the BCR-ABL fusion gene on the Philadelphia (Ph) chromosome, which produces the chromosomal translocation t(9; 22). Although the expression of BCR-ABL can be reduced to low or undetectable levels in some patients who respond well to imatinib, blastic transformation is a major challenge following tyrosine kinase inhibitor (TKI) treatment for a minority of CML patients.
1
This suggests a dismal clinical prognosis, while patients in the chronic phase of chronic myeloid leukemia
Case report
This study was approved by the Institutional Ethical Committee of Lanzhou University Second Hospital (2019A-329). A 51-year-old female was admitted to our hospital in January 2018 because of an 8-month history of left upper abdominal pain. She was diagnosed with CML and received TKI treatment, but progressed to CML-BC 3 months later. Physical examination after admission showed splenomegaly 3 cm under the ribs. Routine blood analysis showed a white blood cell (WBC) count of 306 × 109/L, a hemoglobin count of 107 g/L, and a platelet count of 218 × 109/L, with a classification of 83% neutrophils, 6% lymphocytes, 3% monocytes, 4% basophils, and 4% eosinophils. Basophils were 12.94 × 109/L and eosinophils were 12.15 × 109/L. Biochemistry analysis revealed r-glutamyl transferase 122 U/L and lactate dehydrogenase (LDH) 897 U/L. Regarding bone marrow cell morphology, extremely active proliferation of nucleated cells was evident in the bone marrow and granulocyte proliferation was active; the stage before neutrophilic myelocytes in the bone marrow was dominant, and the number of eosinophilic and basophilic granulocytes had increased compared with normal levels. Karyotype examination on admission (Figure 1) revealed 46, xx, t(9,22)(q34,q11) [20]; the patient was positive for the p210 BCR-ABL gene (gene quantification, 108.91%). Her Sokal score was 0.8. CML-CP was diagnosed and she was treated with imatinib mesylate (400 mg/day).

Twenty metaphase phases were analyzed at the initial diagnosis, revealing a karyotype of 46, xx, t(9; 22)(q34; q11)[20].
Around 2 weeks after initiating treatment, routine blood analysis showed a WBC count of 4.7 × 109/L, a hemoglobin count of 105 g/L, and a platelet count of 275 × 109/L; splenomegaly had disappeared, suggesting the patient was in a stable condition, and complete hematological remission was achieved. She was re-admitted to our hospital in April 2018 because of fatigue and nausea for 8 days and nasal bleeding for 1 day. Routine blood analysis showed a WBC count of 258.43 × 109/L, a hemoglobin count of 98 g/L, a platelet count of 24 × 109/L, 16% neutrophils, 0% lymphocytes, 0% mononuclear cells, 0.01% basophils, and 0% eosinophils. Her basophilic granulocyte count was 1.63 × 109/L and her eosinophil count was 0.21 × 109/L. Biochemical analysis revealed γ-glutamyl transferase 318 U/L and LDH 7742 U/L. Regarding bone marrow cell morphology, bone marrow hyperplasia was highly active with 57.5% of primitive cells, relatively fewer lymphocytes, and an increased number of monocytes; CML was considered. Bone marrow biopsy showed a proliferation of granulocytes, erythrocytes, and mesangium, a proliferation of juvenile cells, focal distribution, a small amount of fibrous tissue hyperplasia, and blast crisis of CML. Flow immunotyping showed that the primitive naive cells accounted for 59.86% of nucleated cells, expressing CD13, CD33, CD34, HLA-DR, myeloperoxidase, and CD38, partially expressing CD117, and weakly expressing CD64. Karyotype analysis was repeated (Figure 2) to show 46, xx, t(2; 5)(p24; q13) [2]/46, idem, t(8; 21)(q22; q22) [5]/47, idem, +8, t(8; 21)(q22; q22) [4]/47, idem, +8, t(8; 21)(q22; q22), and t(9; 22)(q34; q11) [9]. Fifty fusion genes were found, positive quantification of AML1-ETO was 5.65%, and BCR-ABL(P210) positive quantification was 56.96%; thus, CML-BC was considered. On April 16, 2018, the patient suffered a sudden cerebral hemorrhage, family members decided to discontinue treatment, and she died.
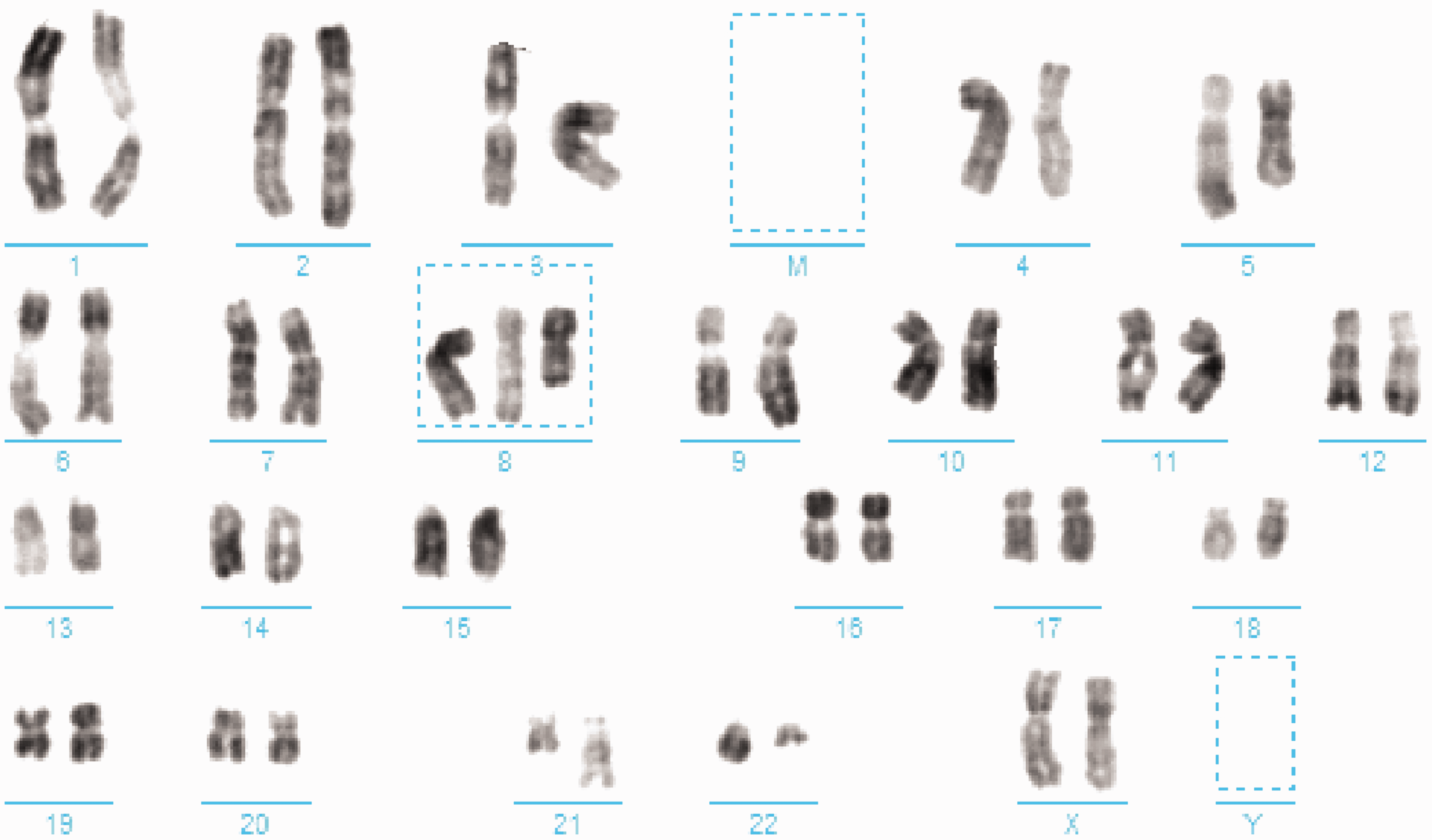
Twenty metaphase phases were analyzed 3 months after treatment. Two showed a chromosome type of 46, xx, t(2; 5)(p24; q13)[2]; five showed a chromosome type of 46, xx, t(8; 21)(q22; q22); four showed a chromosome type of 47, xx, +8, t(8; 21)(q22; q22) [4]; and nine showed a chromosome type of 47, xx, +8, t(8; 21)(q22; q22), t(9; 22)(q34; q11)[9].
Discussion
The prognosis of CML is determined by cytogenetic and molecular abnormalities. The AML1-ETO fusion gene results from the t(8; 21)(q22; q22) chromosome translocation, which is considered to be associated with a favorable prognosis in acute myeloid leukemia (AML-M1). However, ACAs and fusion genes may also determine the course of disease. Moreover, disease progression can occur at any stage through the presence of subclones. Although BCR-ABL is the driving force of disease progression because its reduction is linked to clinical outcomes by TKI treatment, 5 individual BCR-ABL cannot contribute to acute disease.
The latest genetic research shows that one or more mutations is associated with myeloid malignancies, and it is thought that additional mutations besides BCR-ABL can accelerate disease progression. 2 The core binding factor (CBF) of leukemias is t(8; 21)(q22; q22)/AML1-ETO, representing class-II mutations thought to link AML with recurrent genetic abnormalities. These class-II mutations are assumed to cooperate with other mutation types that promote cell proliferation (‘class-I mutations’), such as those in FLT3, RAS, KIT, and JAK2 genes. 3 , 6 According to these two hypotheses, most acute leukemias are the consequence of a collaboration of several types of mutations, which is similar to the pathogenesis of the blast crisis of CML. The BCR-ABL mutation is a common class-I mutation which promotes abnormal cell proliferation. Nevertheless, the downregulation of BCR-ABL gene expression together with AML1-ETO gene positivity is rare in CML-BC, and the poor prognosis of this disease suggests it is caused by a mutation in BCR-ABL. 2 , 7 Hence, the BCR-ABL gene quantification decreased in our patient but CML-CP progressed to BC, which was not consistent with the pathogenesis of CML-BC following effective TKI treatment.
In CML-BC, a newly acquired structural transformation other than a BCR-ABL translocation, including mutations, structural variants, and copy number abnormalities, occurs in the majority of cases of BC. Additionally, more than one cytogenetic abnormality may be involved. 2 Only after the quantitative downregulation of the BCR-ABL gene in the present patient did CML-CP evolve to CML-BC with the appearance of AML1-ETO gene positivity, and little is known about existing clone evolution. The clonal evolution mechanism of the CML mutation may be related to the dominant cloning caused by the proliferation of subclones. Current molecular biology research suggests that tumor evolution in disease progression may be caused by mutations in tumor clone-specific genes, with changes documented in tumor genes, chromosomes, and immunophenotypes; however, tumor clones still persist. 8
Five cases of secondary AML, except CML-BC, have previously been reported, suggesting that Ph+ is a secondary aberration. Similarly, the quantitative downregulation of the BCR-ABL gene along with AML1-ETO positivity in existing CML-BC subclones is rare. This was reported in our CML-CP patient initially in a median risk cytogenetics group, who eventually died. It is unclear whether these subclones were apparent at the initial diagnosis or appeared during disease progression. In the case of our patient, we believe that the existing subclone appeared during the change from CML-CP to CML-BC, similar to other secondary AMLs. This is because she had a good response to earlier TKI treatment, as seen by the downregulation of over 50% of BCR-ABL gene expression. Conversely, in rare cases of CML appearing as de novo BC or rapidly progressing to advanced phases, the Ph translocation was concurrently seen with an inv(16)/CBFB-MYH11. 9
Recently, Singh et al. 10 suggested that analysis of copy number variations, expression profiling, and genome-wide scoring of single nucleotide polymorphisms in different phases of imatinib-treated CML will help our understanding of the resistance mechanism to TKIs. Early use of TKIs was reported to significantly reduce the copy number of BCR-ABL and inhibit the primary clone, while the gradual emergence of subclones was seen with increasing TKI resistance. 11 It is conceivable that TKI resistance is related to subcloning.
In a retrospective study of 2,326 CML patients, Gong et al. 12 , 13 reported that +8 is a major route for ACA, suggesting a good prognosis in the early stages of disease or during therapy. The effect of the prognosis of ACAs will only be reduced if the disease progresses to BC, regardless of the category or complexity of ACAs. Furthermore, the impact of ACA prognosis is related to disease type and stage, and ACAs play an important role in accelerating BC.13–15 However, little is known about how each ACA promotes to BC in the TKI era.