
Abstract
Background:
Hereditary haemorrhagic telangiectasia is an autosomal dominant genetic disorder characterized by abnormalities in blood vessel formation. The clinical manifestations of patients affected with hereditary haemorrhagic telangiectasia include mucocutaneous telangiectasias and visceral arteriovenous malformations.
Case Summary:
We report the case of a 30-year-old female diagnosed with hereditary haemorrhagic telangiectasia presenting with the classic triad of recurrent epistaxis, mucocutaneous telangiectasias and family history of hereditary haemorrhagic telangiectasia with activin receptor-like kinase 1 mutation. Upon skin examination, she was noted to have telangiectasias under left naris, inner lower lip and surface of the tongue, and a vascular malformation on the right forearm.
Conclusion:
Although the skin involvement and epistaxis may be mild symptoms and signs of hereditary haemorrhagic telangiectasia, timely recognition of these can ensure vigilant monitoring of potential severe complications from cerebral and pulmonary visceral arteriovenous malformations.
Background
Hereditary haemorrhagic telangiectasia (HHT), also known as Osler-Weber-Rendu syndrome, is a rare autosomal dominant disorder affecting 1.4 million individuals worldwide.1,2 The mucocutaneous and visceral manifestations of HHT are the result of abnormalities in vascular structure.2,3 Due to lack of intervening capillaries, telangiectasias, arteriovenous malformations (AVMs) or aneurysms can develop in various organs. Although epistaxis and mucocutaneous telangiectasias are often the initial manifestations of HHT, gastrointestinal tract, pulmonary organs or central nervous system can also be affected resulting in serious complications. 4
Recently, four major genes have been implicated in the underlying mechanism of HHT: ENG (endoglin), ACVRL1 (activin receptor-like kinase 1), SMAD4 (mothers against decapentaplegic homolog 4) and GDF2 (growth differentiation factor 2). 5 Mutations in these genes interfere with TGF (transforming growth factor)-beta signalling pathways in vascular endothelial cells, which disrupt cell formation and lead to AVMs. 6 In 2000, Curaçao Diagnostic Criteria was established, and since then, it has been used as the mainstay of HHT diagnosis. The criteria list four characteristics to guide the likelihood of HHT diagnosis: spontaneous recurrent epistaxis, multiple telangiectasias, visceral involvement and family history (see Table 1). 7 The differential diagnosis of HHT includes ataxia-telangiectasias, Bloom Syndrome, CREST syndrome, Fabry Disease, generalized essential telangiectasias, Rothmund syndrome and spider angiomas. 6
Curaçao diagnostic criteria for hereditary haemorrhagic telangiectasias.

Despite the recent advancements in understanding the genetic mechanisms and establishing the diagnostic criteria, diagnosis of HHT is often delayed. 8 It is estimated that one-third of patients wait 1–5 years and 15% of patients wait 6 years or more for a correct diagnosis. 9 Timely diagnosis is essential for preventing and managing visceral complications and promoting adequate genetic testing and counselling for patients and families. Here, we describe a case of HHT with mucocutaneous involvement, chronic epistaxis and a positive family history due to ACVRL1 gene mutation.
Case summary
A 30-year-old female with HHT and a positive family history presented to our dermatology clinic for evaluation of her mucocutaneous papules. On examination, she was noted to have several telangiectasias. One telangiectasia was present under her left naris, one on the inner lower lip, and multiple on the surface of the tongue (see Figures 1–3). No telangiectasias were present on her fingertips, trunk or lower limbs. Physical examination also revealed an erythematous confluent reticulated patch on patient’s right forearm, demonstrating a congenital capillary vascular malformation with no evidence of warmth or thrill (see Figure 4). With regard to this lesion, she complained of pain and darkening of colour during cold temperatures.

Telangiectasias on left naris.

Telangiectasias on surface of the tongue.
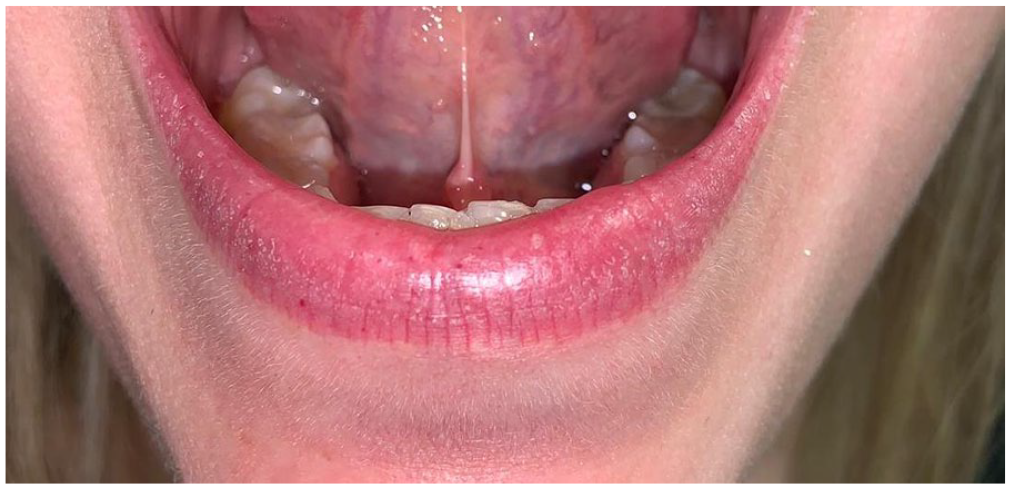
Telangiectasias on inner lower lip.

Vascular malformation on right forearm.
Since the age of 2, the patient had experienced daily epistaxis lasting approximately 10 min and occurring more frequently in dry weather. Her relevant past medical history was positive for anaemia, syncope, pre-menstrual dysphoric disorder managed with fluoxetine as needed and headaches with pressure changes alleviated with acetaminophen. She did not consume any meat products, iron supplements, or any other medications. She had not experienced any rectal bleeding, seizures or shortness of breath.
A review of her family history indicated that the patient’s father and paternal uncle were diagnosed with HHT, and the pathogenetic alteration in the ACVRL1 gene confirmed their diagnosis. In addition, her 2-year-old daughter had been experiencing frequent epistaxis. Thus, the patient was diagnosed with HHT due to the presence of classic triad: recurrent chronic epistaxis, multiple telangiectasias and positive family history. Her investigation included a brain magnetic resonance imaging (MRI) which ruled out AVMs. She was also evaluated at the genetic medicine clinic. The patient agreed to pursue genetic testing to confirm the diagnosis and the underlying genetic mutation, and she was referred to the HHT clinic for a full work-up (i.e. chest x-ray, complete blood count and guaiac stool screen) and ongoing follow-up.
Discussion
This case expands our understanding on the clinical presentation of HHT. Our patient had experienced the first episode of epistaxis at 2 years of age. Approximately, 50% of individuals diagnosed with HHT report epistaxis by 10 years of age and 80%–90% by 21 years. 10 Moreover, up to 95% of patients report developing recurrent epistaxis, similar to our patient. 3 HHT-related epistaxis is caused by rupturing of telangiectasias present in abundance in the nasal mucosa. 3 Epistaxis may be linked to many factors, such as changes in diet, external temperature, humidity, activity and posture.11,12 Specifically, dry weather increased the frequency of epistaxis for our patient. Frequent epistaxis can lead to anaemia, while menorrhagia in female HHT patients may further exacerbate iron deficiency, as seen in our patient. 12
In addition, our case had a family history of ACVRL1 gene mutation, corresponding to HHT2 subtype. There are four subtypes of HHT, each correlating to a specific autosomal gene mutation in ENG, ACVRL1 or SMAD4 (see Table 2). 3 HHT1 and HHT2 are associated with mutation in ENG and ACVRL1 respectively, while the genetic contributor of HHT3 remains unidentified. 13 JP-HHT is a juvenile polyposis-HHT overlap syndrome that accounts for approximately 1% of HHT cases and is due to pathogenic sequence variants in SMAD4. 10 These subtypes display significant differences. For example, pulmonary and cerebral AVMs are more common in HHT1 patients, while hepatic AVMs are more common in those with HHT2 subtype. 10 Thus, it is important to conduct genetic testing and continuously monitor our patient for any signs of hepatic AVMs, if positive for ACVRL1 gene.
Genetic heterogeneity in hereditary haemorrhagic telangiectasias.

HHT: hereditary haemorrhagic telangiectasias; JP-HHT: juvenile polyposis-hereditary haemorrhagic telangiectasias.
AVM has also been described in another genetic syndrome, capillary malformation-arteriovenous malformation (CM-AVM), which is associated with either RAS P21 Protein Activator 1(RASA1) or Ephrin type-B receptor 4(EPHB4) gene mutations. 5 CM-AVM syndrome caused by EPHB4 or RASA1 mutations has similarities in presentation to HHT. As with our patient, they can also lead to telangiectasias and epistaxis. 5 Patients with RASA1-related disorders often present with cutaneous zones of several pinpoint telangiectasias. 5 CM-AVM and HHT may overlap clinically, as the EPHB4 mutation is also associated with HHT. 5
Although the mucocutaneous manifestation of HHT may be considered mild for patients, timely recognition of these skin manifestations can ensure vigilant surveillance of serious complications due to pulmonary and central nervous system AVMs. Embolic cerebral events and transient ischaemic attacks occur in patients with clinically silent pulmonary AVMs and can result in significant morbidity and mortality, indicating the need for early diagnosis and intervention. 3 Given the high misdiagnosis rate, increasing the clinical awareness of HHT for dermatologists can play a significant role in the timely referral of these patients and their family members to receive appropriate preventive care. Due to the systemic nature of this disease, HHT patients should be referred to a multidisciplinary team involving an otolaryngologist, pulmonologist, interventional radiologist, neurologist, geneticist, cardiologist, gastroenterologist, dermatologist, hepatologist and haematologist to provide optimal patient care. Nosebleeds are managed with humidification, nasal lubricants, haemostatic products, laser ablation, sclerotherapy, nasal closure, and oral or topical medications, while iron replacement therapy, surgical resection of bleeding sites and medical therapy can be used to treat gastrointestinal (GI) bleeding. 5 Liver transplantation is recommended for patients with symptomatic hepatic AVMs that cannot be managed medically. 5
Conclusion
Our review highlighted a case of HHT with mucocutaneous telangiectasias, recurrent epistaxis and a positive family history of HHT due to ACVRL1 gene mutation. Although the skin involvement and epistaxis may be mild symptoms and signs of HHT, timely recognition of these symptoms can ensure monitoring of potential complications from cerebral and pulmonary AVMs. Since there are currently no guidelines available regarding the frequency and choice of imaging modality for surveillance of visceral AVMs, further research is warranted.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Ethical approval
This research was conducted ethically in accordance with the World Medical Association Declaration of Helsinki. Written informed consent was obtained from the included patient.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.