
Abstract
The concomitant presence of the XYY syndrome with haematological malignancies is rare. This report presents a case of acute promyelocytic leukaemia (APL) with the promyelocytic leukaemia-retinoic acid receptor alpha (PML-RARA) gene insertional translocation and a chromosome 21 abnormality in a 29-year-old XYY male patient. Karyotype analysis revealed an abnormal karyotype of 47,XYY [14]/46,XYY,–21[16]. Fluorescence in situ hybridization and reverse transcription–polymerase chain reaction analysis showed the existence of a PML-RARA fusion gene. The patient was treated by all-trans retinoic acid (ATRA) and chemotherapy. Laboratory results revealed that the coagulopathy improved and the patient achieved complete remission, based on bone-marrow morphology. The patient then received sequential monthly therapy using arsenic trioxide, followed by ATRA, followed by chemotherapy; he has survived disease-free for 36 months. Our findings suggest that the additional chromosomal abnormalities involving the sex chromosomes and chromosome 21 did not affect the prognosis of APL, and that the sequential treatment strategy had a good clinical effect without being associated with severe side-effects.
Keywords
Introduction
The male sex chromosome aneuploidy 47,XYY is common, with a prevalence ranging between 14.2 and 375 persons per 100000 in newborn boys; it is associated with tall stature, verbal learning disabilities and attention deficits.1,2 Behavioural features described in XYY syndrome include increased risk of impulsivity and difficulties related to behavioural deregulation. 3 In contrast to 47,XXY Klinefelter syndrome, boys with XYY syndrome have normal pubertal development and testosterone levels. 4
Acute promyelocytic leukaemia (APL) is a specific subtype of acute myeloid leukaemia. The t(15;17)(q22;q21) translocation is found in 98% of patients with APL, 5 which reflects the molecular rearrangement of the promyelocytic leukaemia (PML) gene. Additional chromosomal abnormalities (ACA) can also be found in patients with APL in addition to t(15;17)(q22;q21).6,7 The prognostic significance of ACA in APL is unclear to date, but ACAs can involve any human chromosome. To the best of our knowledge, there have been no previous reports on the coexistence of XYY syndrome with APL. The present case report describes a man with XYY syndrome diagnosed with APL with a PML-RARA insertional translocation and chromosome 21 abnormalities.
Case report
A 29-year-old male was admitted to the Department of Haematology, The Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, Guangdong Province, China in July 2011 complaining of repeated fever that had lasted for 2 weeks. His height was 180 cm and weight was 80 kg. He had normal intelligence and did not exhibit aggressive behaviour. Physical examination showed that he had moderate anaemia and multiple petechiae on the bilateral thighs and buttocks, but without superficial lymphadenopathy or hepatosplenomegaly. His peripheral blood count was as follows: white blood cell (WBC) count 0.5 × 109/l; red blood cell count 2.63 × 1012/l; haemoglobin 85 g/l; and platelet count 17 × 109/l. The results of a disseminated intravascular coagulation (DIC) test were as follows: prothrombin time 18.5 s (reference value 11–14.5 s); fibrinogen 1.0 g/l (reference value 2–4 g/l); active partial thromboplastin time 40.6 s (reference value 28–40 s); thrombin time 17.1 s (reference value 14–21 s); D-dimer >20 µg/ml (reference value 0–0.5 µg/ml); plasma protamine paracoagulation test (+). He had normal testosterone levels and had been married for 5 years but had no offspring. Hepatic and renal functions were normal and B ultrasound showed no abnormality in the liver, spleen or kidney. His parents and his brother were healthy.
The blast percentage and assessment of maturation degree were determined by performing a 200-cell leucocyte differential count on peripheral blood smears and a 500-cell differential count on bone-marrow aspirate smears stained with Wright-Giemsa stain. Cytochemical tests included myeloperoxidase (POX) reaction, periodic acid-Schiff (PAS) reaction and nonspecific esterase (NSE) reactions. The bone-marrow characteristic showed marked hyperplasia and the granulocytic cell/erythrocytic cell ratio was 40.7. The granulocytic series accounted for 87.5% of the cell population with 68.5% of promyelocytes and 14.5% of myeloblasts. Promyelocytes were of uneven cell size with irregular nuclear shapes, including being cupped, distorted, folded, segmented and binucleate. The nucleoli were obvious and the cytoplasm was filled with pink granules. Cells occasionally had visible Auer rods and cytoplasmic projections. The erythroid lineage showed normal morphology but was severely inhibited by a proportion of 2% in the sample. Lymphocytes of normal morphology were obviously reduced. Megakaryocytes in the whole slide could not be found; and platelets were widely distributed and extremely rare (Figure 1). Cytochemically, the blasts were negative for PAS and NSE and positive for POX.
Analysis of a bone-marrow smear from a 29-year-old male who presented with repeated fever that had lasted for 2 weeks revealed the replacement of normal bone-marrow cells by promyelocytic leukaemia cells (Wright-Giemsa stain). The colour version of this figure is available at: http://imr.sagepub.com. Scale bar 10 µm.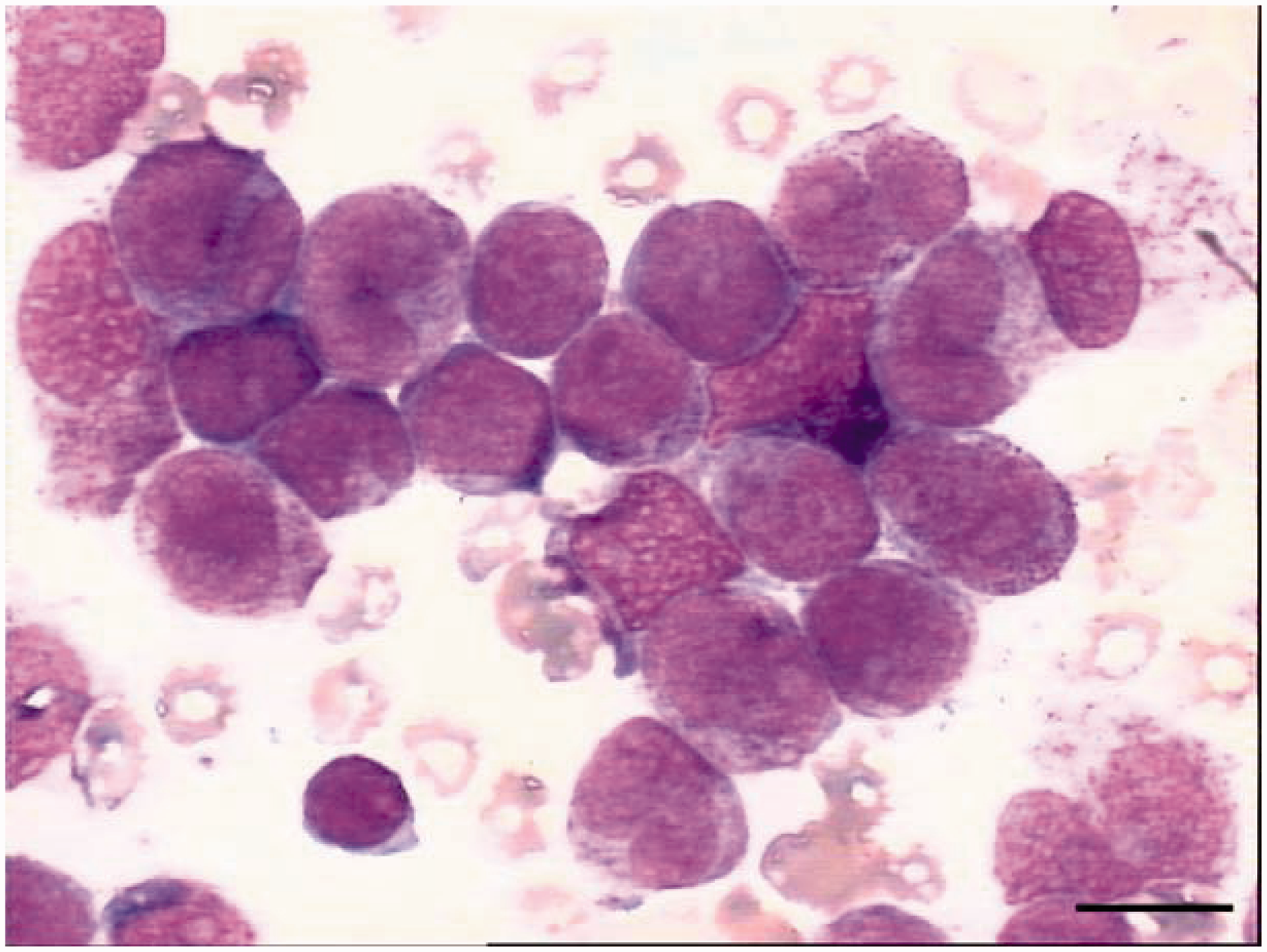
Bone-marrow samples were cultured for 24 h and peripheral blood samples were cultured for 72 h at a concentration of 1–2 × 106 cells/ml, following standard procedures. R band was performed, and chromosomes were identified and analysed according to the International System of Human Nomenclature (ISCN 2005), which defines the karyotype.
8
Cells harvested from the bone-marrow sample were dropped onto four microscope slides. Karyotypes were determined as follows: 47,XYY [14]/46,XYY,–21[16] and did not show t(15;17)(q22;q21) (Figure 2).
Karyotype analysis of a bone-marrow sample from a 29-year-old male demonstrated 47,XYY[14]/46,XYY,–21[16] chromosomal abnormalities. Aberrant chromosomes are indicated by arrows.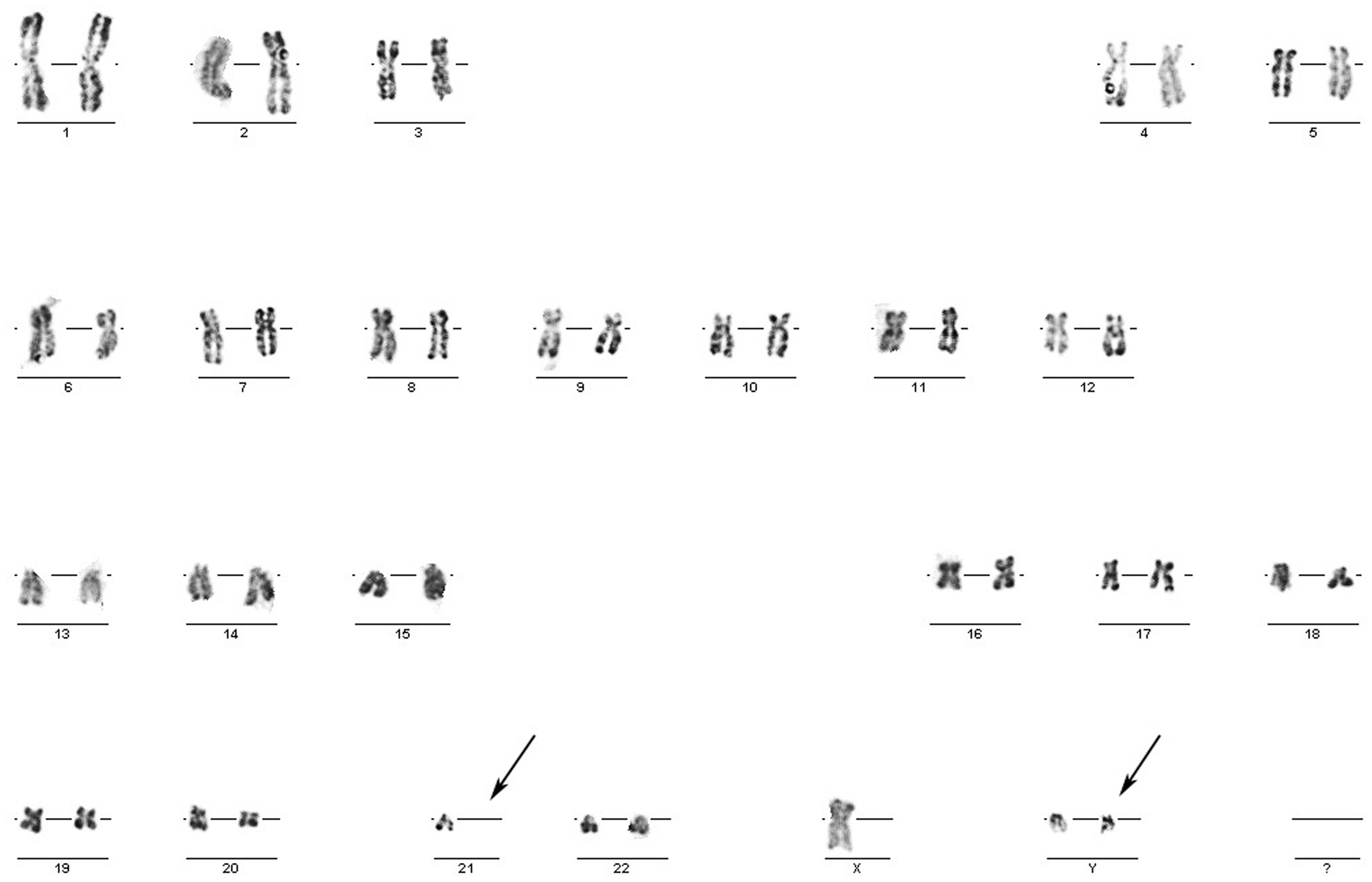
Bone-marrow aspirate was labelled by a panel of directly conjugated monoclonal antibodies (Becton Dickinson, Franklin Lakes, NJ, USA) including antihuman cluster of differentiation CD2, CD7, CD10, CD19, CD20, CD13, CD33, CD14, CD34, CD117 and human leucocyte antigen HLA-DR antibodies. Flow cytometric analyses were performed on a BD FACSCalibur™ system with BD CellQuest™ software (Becton Dickinson). Abnormal cell groups with high-side scatter that resembled granulocytes were observed. Immunophenotypic analysis of the blasts demonstrated the following: positive for CD13, CD33, and CD117; negative for CD34, HLA-DR, CD2, CD7, CD10, CD19, CD20, and CD14.
Fluorescence in situ hybridization (FISH) was performed to analyse the PML-RARA gene rearrangement involving the t(15;17)(q22;q21) and the sex chromosome abnormalities. Dual colour-dual fusion PML/RARA and X/Y centromere probes were used (Cytocell, Cambridge, UK). The PML/RARA DNA probe hybridizes to chromosome 15q22 (spectrum red for the PML gene) and 17q21.1 (spectrum green for the RARA gene), so the fusion gene is yellow (Y). The X chromosome is marked in green (G) and the Y chromosome in red (R), so the normal female has a 0R2G signal pattern and the normal male displays a 1R1G signal pattern. Briefly, the sample of bone marrow was dropped onto glass slides and air-dried. The slides were immersed in 2× saline sodium citrate (SSC) at 37℃ for 30 min, then rinsed in a graded series of ethanol (70%, 85% and 100%, each concentration was applied for 2 min) at room temperature and air-dried. Slides were hybridized for 16 h at 37℃ in a humidified chamber. After that, slides were washed in 0.4× SSC/0.3% NP-40 for 2 min, then in 2× SSC/0.1% NP-40 for 30 s. Slides were counterstained with 0.125 µg/ml 2 -(4-amidinophenyl)-6-indolecarbamidine dihydrochloride and analysed under fluorescence microscopy. A total of 200 interphase cells were analysed and 90% of them had a 2R1G1Y signal pattern, implying a PML-RARA insertional translocation (Figure 3A); this was confirmed by metaphase FISH analysis (Figure 3B). When the X/Y probe was used, 100% cells had a 2R1G signal pattern, indicating that cells from the patient had two Y chromosomes (Figure 4). In order to rule out whether the additional Y chromosome was associated with leukaemia, karyotypes of peripheral blood cells were analysed during the complete remission period. They were found to have 47,XYY, which confirmed that the abnormal Y chromosome was caused by congenital anomalies.
(A) Fluorescence in situ hybridization (FISH) analysis using a PML/RARA DNA probe on interphase cells. PML gene probe (15q22) was labelled with spectrum red; RARA gene probe (17q21.1) was labelled with spectrum green. One green signal (RARA gene), two red signals (PML gene) and one yellow signal (fusion gene) were noted. Scale bar 10 µm. (B) FISH analysis with the same probe on metaphase cells. The colour version of this figure is available at: http://imr.sagepub.com. Scale bar 10 µm. Fluorescence in situ hybridization analysis using X/Y centromere probes. X chromosome is labelled green (G); Y chromosome is labelled red (R). The normal female has a 0R2G signal pattern and the normal male has a 1R1G signal pattern. 2R1G signals were observed, indicating the addition of a Y chromosome. The colour version of this figure is available at: http://imr.sagepub.com. Scale bar 10 µm.

Total cellular RNA was extracted by standard laboratory procedures from fresh bone marrow cells using TRIzol® reagent (Invitrogen, Carlsbad, CA, USA). Reverse transcription of the RNA to cDNA was undertaken using a Maxima™ First Strand cDNA Synthesis Kit (Fermentas, Glen Burnie, MD, USA). Primers for the PML-RARA fusion gene were designed and detected as previously described.
9
Products of the reverse transcription–polymerase chain reaction (RT–PCR) were separated on 2% agarose gel and analysed using an ultraviolet transilluminator. Only the L-form (427 base pairs) of the PML-RARA transcript was amplified by RT–PCR, indicating that the patient carried the L-form of the PML-RARA fusion gene, as described in another study on acute leukaemia (Figure 5).
9
Detection of the PML-RARA fusion gene transcript by reverse transcription–polymerase chain reaction. Lane M: molecular weight markers (base pairs [bp]); lane S: S-form of PML-RARA transcripts were negative; lane L: L-form (427 bp) of PML-RARA transcripts were amplified; lane N: negative control; lane B: blank control.
After the diagnosis of APL was confirmed, the patient was treated with 20 mg all-trans retinoic acid (ATRA), orally, twice daily for 1 week. The patient also received 2000 AxaIU low molecular weight heparin calcium, twice daily by subcutaneous injection for 6 days, and an infusion of fresh plasma to prevent DIC. The WBC count increased to 12 × 109/l and blood coagulation function returned to normal after treatment for 1 week. Induction chemotherapy was carried out as follows: 40 mg darubicin intravenous (i.v.) injection once a day for 3 days; 100 mg cytosine arabinoside i.v. infusion twice a day for 7 days (known as the DA regimen). The patient achieved complete remission after this first course of chemotherapy. Consolidated treatments consisted of three regimens: 10 mg arsenic trioxide i.v. infusion once daily for 14 days in the first month; 20 mg ATRA administered orally twice daily for 28 days in the second month; chemotherapy (see below) in the third month. These three-monthly regimens were carried out in sequence. In the first year, these regimens were repeated four times over the 12-month period. In the second and third years of treatment, the three regimens were scheduled to be administered in the same sequence, but with a 1-month treatment-free period between each monthly treatment. The chemotherapy regimens that were used in the third month comprised of either the DA regimen or 10 mg idarubicin i.v. injection once daily for 3 days and 100 mg cytosine arabinoside i.v. infusion twice daily for 7 days (known as the IDA regimen); or 12 mg mitoxantrone i.v. infusion once daily for 3 days and 100 mg cytosine arabinoside i.v. infusion twice daily for 7 days (known as the MA regimen); or 40 mg pirarubicin i.v. injection once daily for 3 days and 100 mg cytosine arabinoside i.v. infusion twice daily for 7 days (known as the TA regimen).
The patient achieved complete remission following the first course of treatment and has received eight courses of chemotherapy, arsenic trioxide and ATRA during consolidated treatment, to date. The patient has survived disease-free for 36 months and has remained in complete remission. After complete remission was achieved, a repeated cytogenetic analysis of both bone marrow and peripheral blood showed the continued presence of 47,XYY without any evidence of the chromosome 21 abnormality, indicating that the XYY chromosome aneuploidy was unrelated to the APL.
Discussion
Trisomy 21, also known as Down’s syndrome, is associated with an increased incidence of cancer. 10 In particular, children with Down’s syndrome have an increased risk of leukaemia. 11 The natural history of leukaemia in children with Down’s syndrome suggests that trisomy 21 and GATA1 genetic mutations contribute to the malignant transformation of haematopoietic cells. 12 Like Down’s syndrome, XYY syndrome is a chromosomal abnormality that is often associated with cancer. 1
A few haematological malignancies have been reported in patients with XYY syndrome, occurring in acute myeloid leukaemia without maturation, acute myeloid leukaemia with maturation, chronic myeloid leukaemia, and acute lymphoblastic leukaemia.13–17 To the best of our knowledge, this is the first case reporting a patient with 47,XYY syndrome with APL. The male patient described in this current case report had a tall stature, normal intelligence and testosterone levels, and did not exhibit aggressive behaviour. Cytogenetic analyses of a pretreatment bone-marrow sample, and postremission samples of bone marrow and peripheral blood, all showed 47,XYY, thus XYY syndrome was confirmed.
According to World Health Organization guidelines, 18 the diagnostic criteria for APL state that the proportion of abnormal promyelocytes must be >20% by bone-marrow cell morphological appearance. However, if t(15;17)(q22;q21) is observed, even if the proportion of abnormal promyelocytes is <20%, the patient can still be diagnosed with APL. In this present case, abnormal promyelocytes accounted for 68.5% of the total cells, although conventional cytogenetic analysis did not show t(15;17)(q22;q21). Immunological examinations showed that the abnormal cell group expressed myeloid markers but was negative for CD34 and HLA-DR, which accords with the typical appearance of APL. In addition, FISH and RT–PCR analysis demonstrated the PML-RARA fusion gene. Taking this evidence together resulted in the patient being diagnosed as having APL.
Conventional cytogenetic analysis is the main method used to test for t(15;17)(q22;q21). However, complicated translocation variations, including a three-way translocation or the PML-RARA insertion translocation, also exist; these cannot be tested by cytogenetic analysis.19,20 These cryptic t(15;17)(q22;q21) rearrangements can be detected by molecular methods, such as FISH. The use of the dual colour-dual fusion PML/RARA translocation DNA probe with high sensitivity and specificity detects nearly all of the PML-RARA fusion signal of cryptic t(15;17)(q22;q21) rearrangements. In this current case report, karyotype analysis did not show the t(15;17)(q22;q21) rearrangement. However, the interphase FISH analysis showed a 2R1G1Y signal pattern, indicating the PML-RARA insertional translocation, which was confirmed by analysis of the metaphase FISH. RT–PCR analysis confirmed that the L-form PML-RARA fusion gene existed in this patient. These findings suggest that a combination of conventional karyotype analysis and molecular methods will lead to a more comprehensive understanding of genetic changes associated with APL and reduce the PML-RARA omission ratio.
Chromosomal abnormalities in addition to t(15;17)(q22;q21), have been reported in between 26% and 39% of APL cases.6,7 ACA can involve any human chromosome. Trisomy 8 is the most frequent secondary anomaly, although other abnormalities involving chromosomes 1, 3, 7, 9 and 11 have been described.6,21 The prognostic value of additional cytogenetic changes to the t(15;17)(q22;q21) remains uncertain. According to previous reports,22,23 ACA constitutes an unfavourable prognosis. However, several studies have shown that the coexistence of ACA in patients with APL did not affect clinical and biological characteristics, complete remission rate, disease-free survival rate, overall survival rate or relapse rate.7,21 Wiernik et al. 24 reported that treatment with chemotherapy alone did not affect overall survival and disease-free survival rates of patients with ACA, although when using ATRA treatment (with or without chemotherapy) patients who only had t(15;17)(q22;q21) had higher overall survival and disease-free survival rates compared with patients with ACA. These findings suggest that the presence of ACA reduced the sensitivity of cancer cells to the ATRA treatment, and reduced the curative effects. The current case had congenital sex chromosome abnormalities followed by chromosome 21 loss, but he reached complete remission after the first course of chemotherapy and has survived disease-free for 36 months. In the current case, ACA involving the sex chromosomes and chromosome 21 did not appear to affect the outcome of APL, although the patient had received ATRA treatment.
The specific marker for APL, the t(15;17)(q22;q21) translocation, has diagnostic and prognostic significance. In clinical practice, the identification of the t(15;17)(q22;q21) translocation predicts sensitivity to ATRA. 25 The transcription factor PU.1 plays a critical role during myeloid differentiation 26 and abnormal activation of PU.1 is associated with development of myeloid leukaemia. 27 The PML-RARA protein can combine with PU.1 to form a PML-RARA-PU.1 complex, thus inhibiting PU.1 expression and the subsequent activation of the target genes controlling the differentiation of myeloid progenitor cells; this eventually leads to the development of leukaemogenesis. 28 However, ATRA can recover PU.1 expression by binding with the ATRA receptor domain on the PML-RARA protein, which induces the differentiation of leukaemic cells. 29 Arsenic binds directly to cysteine residues in zinc fingers located within the RBCC domain (which includes one RING domain, two B box motifs, and a coiled-coil domain) of the PML-RARA and PML proteins; this induces PML oligomerization, which increases its interaction with the small ubiquitin-like protein modifier. 30 This allows the modified proteins to be degraded by proteasome, resulting in the induction of differentiation and apoptosis of the leukaemia cells. 30 Therefore, the complete remission rate and 5-year disease-free survival rate are both above 90% when ATRA is combined with arsenic trioxide to treat APL. 31
In the authors’ institution, APL is treated using sequential consolidated regimens that consist of arsenic trioxide followed by ATRA, and then chemotherapy. These three monthly regimens are carried out in sequence and repeated over three years, as described above, with a 1-month treatment-free period between each monthly treatment in the second and third years. Using 10 mg/day arsenic trioxide for 14 days had a good effect on leukaemia cells while producing only mild side-effects in the liver and kidney. Chemotherapy was followed by another course of arsenic trioxide to ensure normal recovery of haematopoiesis. Meanwhile, arsenic trioxide and ATRA can lessen the frequency of chemotherapy, which in turn reduces bone-marrow injury. As a result of the fact that the XYY syndrome involves multiple organ dysfunction, toxicity from chemotherapy can be severe.13,14 However, the case described in this current report underwent each course of chemotherapy without experiencing severe side-effects. Therefore, the authors recommend that the sequential strategy is appropriate for patients with XYY syndrome and APL, as it was associated with good clinical outcomes and few side-effects.
Footnotes
Declaration of conflicting interest
The authors declare that there are no conflicts of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.