
Abstract
Acute promyelocytic leukaemia consists of 7%–8% of cases of acute myeloid leukaemia. Extramedullary manifestations are rare and show distinct biological features. We describe a 22-year-old female of Malay ethnicity who presented with fever and a left axillary swelling for a week. The peripheral blood smear showed abnormal promyelocytes with faggot cells. PML-RAR-alpha t(15;17) (q22; q12) was detected by polymerase chain reaction. The left axillary swelling histology and immunohistochemical staining confirmed granulocytic sarcoma. She was induced with triple agents consisting of all-trans-retinoic-acid, arsenic trioxide and idarubicin. On day 14 of induction, she developed severe neutropenic sepsis in which she responded to ventilation and antimicrobials. She completed her induction, consolidation and maintenance therapy. Currently she is in molecular and morphological remission. Extramedullary disease in acute promyelocytic leukaemia usually has a severe clinical presentation. Granulocytic sarcoma may present as an early feature of acute promyelocytic leukaemia.
Introduction
Acute promyelocytic leukaemia (APL) consists of 7%–8% of adult acute myeloid leukaemia cases. 1 APL was first characterised by French and Norwegian physicians as a hyperacute fatal disease with mean survival time of less than a week. 2 However, in modern times, APL has become highly curable with 10-year survival rates approaching 90%. 3 This disease is more commonly seen in children and has distinct clinical characteristics as compared to other subtypes of acute myeloid leukaemia. In children, the disease is commonly associated with leucocytosis (white blood cell (WBC) count > 10,000/uL) which is seen in the microgranular variant of APL and they are susceptible to treatment-related toxicities such as pseudotumour cerebri. 3 These patients may have a high occurrence of disseminated intravascular coagulopathy which show a good response to all-trans-retinoic acid (ATRA). 4 Approximately 10%–25% of adult APL patients may relapse after achieving complete remission. 4 Extramedullary manifestations (EM) in APL are rare at first presentation and show distinct biological features. 5 In all, 3%–5% of patients will suffer extramedullary relapse. 5 The incidence of EM has been rising in the era of ATRA therapy. Some studies have suggested a possible relationship between ATRA therapy and EM. 6 There are two reasons for this. The first is probably attributed to the effect of ATRA on adhesion molecules which may result in increased infiltrative capability of the APL leukemic blasts at the sanctuary sites and the second is due to the prolonged survival in treated APL patients. 6 Despite advances in therapy, high-risk APL is associated with early mortality due to bleeding complications. The Modified Sanz score 2017 subdivides APML into three risk groups: low, intermediate, and high risk with the high risk being defined as WBC more than 10 x 109/L 7 . This score was developed by the Spanish Programme for the Study and Treatment of Haematological Malignancies (PETHEMA) and Italian Adult Haematological Diseases Group (GIMEMA) and it correlates with relapse-free survival (RFS) with more relapse seen in the high-risk group. 7
Case report
A 22-year-old female of Malay ethnicity presented to the haematology department with fever and left axillary swelling for a week. She has no past medical or family history. She is a non-smoker and does not consume alcohol. She works as a bank clerk.
On examination, she was pale. She had a left axillary swelling of 5 x 5 cm (Figure 1(a)) which was firm, non-tender with no discharge. Other systemic examinations were unremarkable with no presence of palpable lymph nodes or organomegaly.

(a) Left axillary mass 5 x 5 cm, (b) chest radiograph reveals diffuse nodular pulmonary infiltrations, and (c) computed tomography of pulmonary arteries shows pulmonary vessels well opacified with no filling defects.
Her complete blood count revealed normochromic normocytic anaemia of 5.5 g/dL with leucocytosis at 22 x 109/L and thrombocytopenia at 30 x 109/L. She had no coagulopathy.
The peripheral blood smear (Figure 2(a)) and bone marrow aspirate (Figure 2(b)) showed presence of blasts, abnormal promyelocytes and faggot cells. The trephine biopsy microscopy (Figure 2(c)) showed a hypercellular marrow which comprised almost entirely of blast population expressing myeloperoxidase (MPO) but lacking CD34 and CD3 on immunohistochemistry (Figure 2(d)–(f)). Marrow flow cytometry revealed 69% cluster of aberrant myeloid cells expressing CD117, cMPO, CD13, CD33, CD38 but lacking CD34 and HL-DR. Marrow for molecular PML-RAR-alpha fusion gene t(15;17) (q22; q12) was detected by polymerase chain reaction (PCR). The left axillary swelling histology showed diffuse monomorphic infiltrates with neoplastic cells exhibiting fine nuclear chromatin and moderate rim of basophilic cytoplasm. On immunohistochemistry, the cells were positive for MPO (diffuse positivity), CD13, CD33, CD68 and CD117. The cells were negative for CD34 and HLA-DR. These findings were consistent with granulocytic sarcoma.
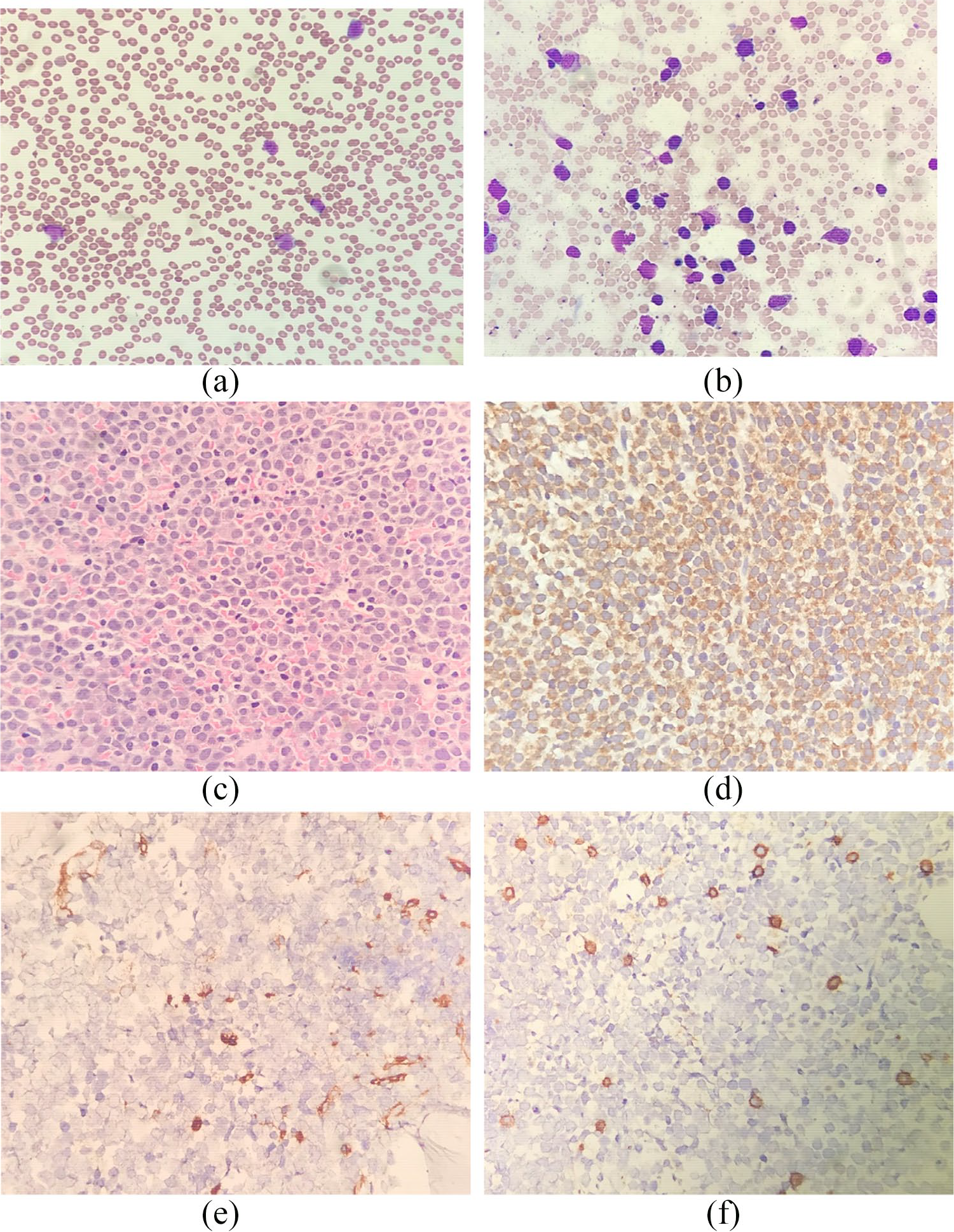
(a) Peripheral blood film shows moderate-to-large sized blasts with bilobed nuclei and presence of faggot cells. (b) Bone marrow aspirate reveals many moderate-to-large sized blasts with prominent nucleoli, bilobed cells and faggot cells. (c) Trephine biopsy microscopy (H&E x 40) shows hypercellular marrow which comprises almost entirely of blasts. (d–f) Panel of immunohistochemical staining (40x magnifications) shows diffuse positivity of MPO, negativity for CD34 and reactive CD3 cells.
She was diagnosed as high-risk APML (Modified Sanz score 2017) with granulocytic sarcoma (GS). She was treated with dexamethasone prophylaxis for differentiation syndrome (DS) of 10 mg twice daily and triple therapy induction comprising of daily oral all-trans-retinoic-acid (ATRA) 45 mg/m2, intravenous arsenic trioxide (ATO) 0.15 mg/kg for a total of 25 doses and 3 doses of intravenous idarubicin. On day 14 of induction, she developed culture-negative neutropenic sepsis. She was in Type 1 respiratory failure. She was intubated and nursed at the intensive care unit. There was no evidence of alveolar haemorrhage. The chest radiograph (Figure 1(b)) revealed diffuse nodular pulmonary infiltrations. The computed tomography of the pulmonary arteries (Figure 1(c)) showed no features of pulmonary embolism. Serum galactomannan was not detected.
She was treated with antimicrobials in which she responded completely. She has completed her induction, consolidation and maintenance therapy. She has been in complete molecular remission for the past 2 years.
Discussion
We describe a case of high-risk APL with extramedullary disease (EMD) in an adult. APL is characterised by the presence of a balanced reciprocal chromosomal translocation involving the retinoic acid receptor-alpha gene on chromosome 17 (RARA). A translocation of t(15;17) is seen in 95% of the cases of APL. 8 There are eight other gene rearrangements which may also occur in the remaining 5% of APL, namely, fusion of RARA to nucleophosmin (NPM1), nuclear matrix associated (NUMA1), promyelocytic leukaemia zinc finger (PZLF), signal transducer and activator transcription 5b, protein kinase A regulatory subunit 1-alpha, BCL6 corepressor (BCOR), factor interacting with PAPOLA and CPSF1. 8
EM may occur at any time along the course of the disease and it is associated with a poorer outcome. 9 EM is considered rare in APL. EM can occur in isolation most frequently in the central nervous system or it can be associated with bone marrow involvement. 9 The other sites of involvement are the skin, testes, lymph nodes, mediastinum, and gingiva. Several factors contribute to a higher incidence of EMD in APL. They are young age < 45, peripheral leucocytosis, microgranular morphology, expression of CD56 and PML-RAR-alpha bcr3 isoform expression. 10
Our patient presented with EM in the form of GS of the left axilla. GS consists of immature and mature myeloid cells and is commonly associated with chronic myeloid leukaemia or de novo acute myeloid leukaemia with myelomonocytic or monocytic morphology (French-American-British subtypes). 11 GS was previously known as chloroma due to its greenish colour attributed to the high content of MPO. 12 GS in APL is rare and only a few such cases have been reported in the literature. Seldomly, GS may precede APL. Those cases reported occurred in a relapsed setting of APL.
Our patient demonstrated Type 1 Respiratory failure on day 14 of induction therapy. The probable causes are neutropenic septicaemia, DS, alveolar haemorrhage and pulmonary embolism. Injudicious use of steroids for DS prophylaxis is a potential cause of infection. DS prophylaxis in this case could mask the signs of infection and impair the natural immune response leading to increased susceptibility to infection. It can, at times, pose a dilemma in differentiating between sepsis and DS as clinical manifestations are often similar in nature for both conditions. Inflammatory markers such as C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) could be elevated in both situations, thus providing no additional value.
DS is a cytokine release syndrome often associated with peripheral leucocytosis at presentation. It is characterised by hypotension, pyrexia, peripheral oedema and multi-organ failure. 13 DS has a bimodal time distribution with the first peak during the first week (47%) and the second peak during the third week (25%). 14 Early severe DS more often required mechanical ventilation and was associated with a higher mortality. 14 Gross myeloid differentiation in the marrow despite peripheral leukopenia could also contribute to DS. Some studies have shown conflicting results in terms of reduction in DS-related complications with steroid prophylaxis in patients presenting with leucocytosis. Wiley and Firkin demonstrated a reduction in DS-related pulmonary complications when prednisolone was given to patients with WBC counts higher than 5 x 109/L. The European APL group suggested that patients with leucocytosis given steroid prophylaxis required less mechanical ventilation. 15 Despite the lack of strong evidence, some major groups have advocated the use of DS prophylaxis. 16 Prophylaxis commonly suggested are oral prednisolone 0.5 mg/kg per body weight per day from day 1 until end of induction therapy, methylprednisolone 20–50 mg/day for 5–10 days or dexamethasone 2.5 mg/m2 per 12 hours (maximum dose of dexamethasone 10 mg/dose) for patients with WBC counts higher than 5 x 109/L 16 . In this case, we used a higher than usual recommended dose of oral dexamethasone of 10 mg twice daily which was the maximum dose used in some studies as we thought it would be more effective in preventing DS without realising its potential cause for infection.
High-risk APL is associated with haemorrhagic and thrombotic concerns. The bleeding diathesis is contributed by several factors namely: overexpression of Annexin 11, low levels of alpha2-antiplasmin, and elevated levels of urokinase plasminogen activator resulting in hyperfibrinolyis. 17 The thrombotic concerns are largely attributed to promyelocytes releasing procoagulants such as tissue factor and cancer procoagulant. Tissue factor is an activator of coagulation, and it is highly expressed in APL cells which play an important role in clotting. 17
ATRA-ATO combination for induction and consolidation has become the standard of care for low to intermediate-risk APL patients. Maintenance therapy can be safely omitted in this group of patients. 18 High-risk APL usually requires a combination of ATRA-ATO and chemotherapy such as idarubicin or an antibody-drug conjugate such as gemtuzumab ozogamicin (GO). The combination of ATRA-ATO and GO is safe and effective in high-risk APL resulting in a complete remission rate of 96%. 18 This chemotherapy-free regimen reduced induction morbidity and spared patients the toxicities associated with cytotoxic chemotherapy especially in the elderly. 18
About 50% of patients treated with ATRA-ATO-based regimens without chemotherapy develop leucocytosis. 19 In patients with marked hyperleukocytosis when ATRA-ATO-based regimens are used, a reasonable option is the administration of cytoreductive drugs such as hydroxyurea, anthracyclines or GO. 19 However, in patients with hyperleukocytosis treated with ATRA-based chemotherapy regimens, very early administration of chemotherapy is recommended besides the use of steroid prophylaxis. 19 Omission of maintenance can be considered if the patient has achieved molecular remission at the end of consolidation in high risk APL. In our patient, we chose to administer idarubicin instead of GO as GO was unavailable at our setting.
Conclusion
In conclusion, our case illustrates a successfully treated young female with high-risk EMD in APL who presented with multiple complications and challenges during her course of treatment. She has been in complete remission for the past 2 years since completion of therapy. Prompt diagnosis, patient’s compliance to therapy, early recognition of complications during therapy, availability of potent differentiating agents, lack of additional adverse cytogenetic and molecular abnormalities and good in-hospital multidisciplinary support could have contributed to her successful outcome. With improvised understanding of the disease biology and mechanisms that underlie high-risk EMD in APL, more efficacious therapy and prophylaxis could be devised to overcome these challenges.
Footnotes
Author contributions
G.K. analysed the data, designed the paper, and contributed to the writing of the manuscript. J.S. made critical revisions and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
Ethical approval is not required as this is not a clinical trial.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Guarantor
Ganesh Kasinathan is the guarantor of this manuscript.