
Abstract
Objective
To investigate the efficacy and safety of 14 days’ orally administered tolvaptan as adjunctive treatment for hepatic oedema in Japanese liver cirrhosis patients with insufficient response to conventional diuretics, with the option to increase dose in those who did not respond initially.
Methods
This multicentre, single-arm, phase 3 study allocated patients with liver cirrhosis and persistent ascites to 7-day treatment with 7.5 mg/day tolvaptan followed by an additional 7 days’ treatment. Responders at day 7 (achieving ≥1 kg body-weight reduction) continued on 7.5 mg/day tolvaptan; nonresponders (<1 kg body-weight reduction) received 15 mg/day tolvaptan. Conventional diuretic treatment continued throughout. The primary endpoint was change in body weight from baseline, as a marker of ascites volume.
Results
A total of 51 patients received 7.5 mg/day tolvaptan for 7 days, which caused a significant reduction in mean body weight (55% response rate). During the second 7-day treatment period, 30 patients received 7.5 mg/day tolvaptan and 13 patients received tolvaptan 15 mg/day: response rates were 43% and 23%, respectively. Two serious adverse events were observed. Serum sodium was within normal range.
Conclusions
Tolvaptan therapy for 14 days (with possible dose increase as necessary), in combination with conventional diuretics, effectively reduced body weight in patients with hepatic oedema.
Introduction
Patients with liver cirrhosis and with hepatic oedema are at high risk of further complications of hepatic disease.1–4 Sodium dietary restriction and diuretic therapy are the primary treatment modalities for patients with hepatic oedema-manifested ascites. 5 Such diuretics often cause electrolyte imbalance before producing a sufficient amount of diuresis, however,6–9 which makes it difficult to make escalating dose adjustments for optimal diuretic effect in patients with hepatic dysfunction.2,10
Tolvaptan (Samsca™; Otsuka Pharmaceutical Co. Ltd, Tokyo, Japan), a novel oral aquaretic agent, is an arginine vasopressin nonpeptide V2 receptor antagonist, developed for the treatment of clinically significant hypervolaemic or euvolaemic hyponatraemia or less marked hyponatraemia in patients with heart failure, cirrhosis or syndrome of inappropriate antidiuretic hormone.11–15 Tolvaptan is indicated for volume overload in heart failure at a daily dose of 15 mg/day, in Japan.16–18 In addition to these indications, add-on therapy of tolvaptan to conventional diuretics is expected to be useful for the treatment of hepatic oedema, since tolvaptan induces the secretion of free water without sodium excretion (aquaresis). 19
In a pharmacokinetics/pharmacodynamics study, tolvaptan was shown to decrease body weight and ascites volume, and increase urine output, in a dose-dependent manner. 20 Additionally, a dose-finding study investigating the effect of a 7-day treatment with tolvaptan 7.5, 15, and 30 mg/day in hepatic oedema, determined that the optimal dose of tolvaptan was 7.5 mg/day, based on reductions in body weight and abdominal circumference. 21 Although doses of tolvaptan ≥15 mg/day have shown an aquaretic effect, 22 the effect of increasing the dose from 7.5 mg/day to 15 mg/day in patients who do not initially respond to 7.5 mg/day has not been investigated. Thus, the present study investigated whether a diuretic effect could be produced by increasing the dose to 15 mg/day for an additional 7 days, in patients who showed insufficient aquaretic effect after an initial 7-day administration of tolvaptan 7.5 mg/day.
Patients and methods
Study population
This was a multicentre, single-arm, phase 3 study conducted between January and December 2010 at 39 study sites in Japan. Consecutive male or female patients with cirrhosis of the liver, who met all of the following criteria, were eligible for inclusion in the study: (i) age 20–80 years; (ii) persistent ascites despite conventional diuretic treatment; (iii) previous imaging diagnosis of liver cirrhosis; (iv) patients whose dose of conventional diuretics could not be increased due to adverse drug reactions or risk of adverse drug reactions, or where efficacy was insufficient; (v) patients who had been receiving one of the following combination therapies (loop diuretic and oral antialdosterone agent from ≥7 days prior to acquisition of informed consent): (a) loop diuretic dose equivalent to ≥40 mg/day furosemide and ≥25 mg/day spironolactone or (b) loop diuretic dose equivalent to ≥20 mg/day furosemide and ≥50 mg/day spironolactone; (vi) inpatients at, or patients who could be admitted to, one of the study sites. Major exclusion criteria were: (i) hepatic encephalopathy (Inuyama classification 23 ≥grade II); (ii) history of cerebrovascular disorder; (iii) pregnancy; (iv) history of treatment with albumin products or blood products containing albumin; (v) patients otherwise judged by the investigator to be inappropriate for inclusion in the study.
Written informed consent was obtained from all patients. The study protocol was approved by the institutional review board at each study site, and the study was conducted in compliance with the Declaration of Helsinki, 24 and according to the principles of Good Clinical Practice. This study was registered on ClinicalTrials.gov (NCT01048788).
Study design
The study consisted of four periods: a 10-day screening period after obtaining informed consent; a 3-day pretreatment observation period (defined as baseline); a 14-day treatment period (the initial 7 days was defined as Treatment Period-I, and the subsequent 7 days as Treatment Period-II); a 7-day post-treatment observation period (Figure 1). For all variables, data obtained immediately before the start of trial-drug administration were used as baseline data. The dose and regimen of conventional diuretics used prior to enrolment of this study were unchanged until the completion of the final assessment on the day following the final administration of tolvaptan. Patients in whom body weight before breakfast on the second and third days of the pretreatment observation period was stable (±1.0 kg) were eligible for advancement to the treatment period, by investigator’s judgement.
Design of a study to assess the efficacy and safety of tolvaptan in patients with hepatic oedema-associated ascites refractory to conventional diuretic treatment. One-way arrow represents the patients with insufficient response after the initial 7 days’ treatment with tolvaptan 7.5 mg/day (Treatment Period-I), who received an increased dose of tolvaptan 15 mg/day in Treatment Period-II.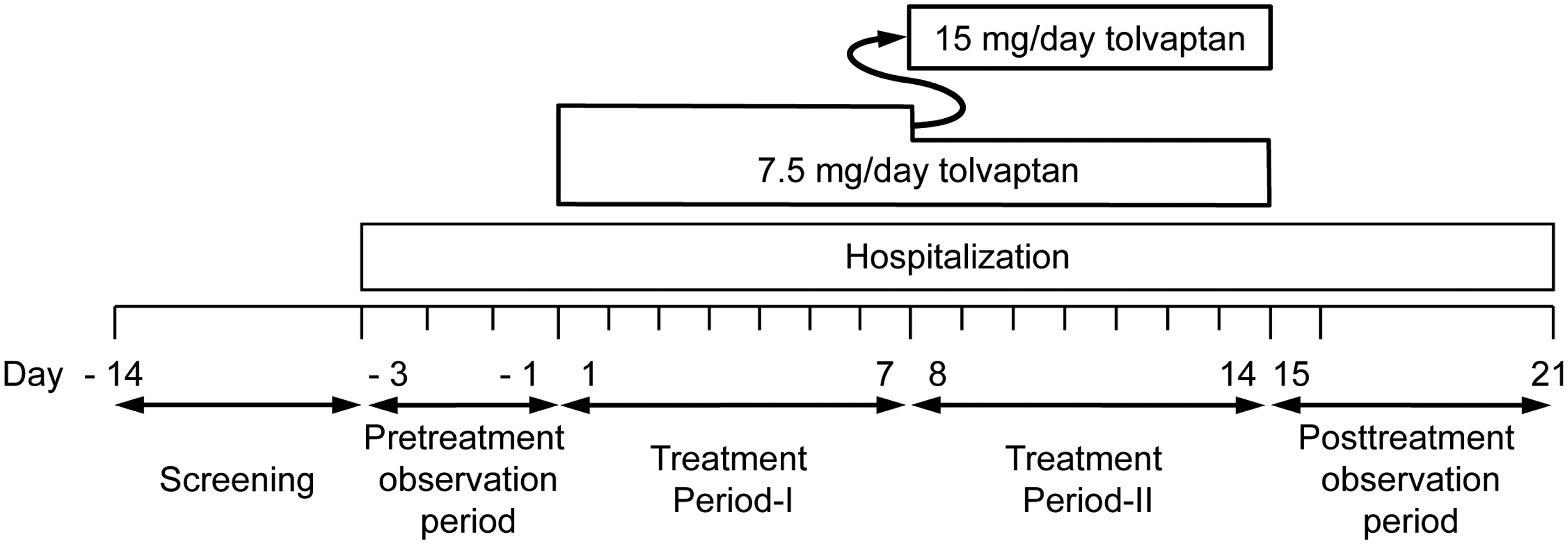
During Treatment Period-I, all patients received 7.5 mg tolvaptan orally, once daily, after breakfast in combination with their fixed regimen of conventional diuretics (as only 15 mg and 30 mg tolvaptan tablets are licensed, 7.5 mg tablets made specifically for experimental use in clinical studies were administered). Patients who achieved a body-weight reduction of ≥1.0 kg by the end of Treatment Period-I continued to receive 7.5 mg/day tolvaptan during Treatment Period-II. Patients who did not achieve ≥1.0 kg reduction in body weight by the end of Treatment Period-I received 15 mg/day tolvaptan in Treatment Period-II. Patients continued to receive 7.5 mg/day tolvaptan during Treatment Period-II if the investigator judged dose escalation to be inappropriate, based on the patient’s volume expansion and any adverse events observed. Patients were hospitalized (for close monitoring) from the day before the pretreatment observational period until completion of the post-treatment observation period.
Efficacy evaluation
Improvement in hepatic oedema was assessed by the reduction in body weight as a marker for decrease in ascites volume, 1 and the primary endpoint was change in body weight from baseline. Body weight was measured every day throughout the treatment period, and abdominal circumference was measured on days 7 and 14, before breakfast and after urination. Patients who showed a body weight reduction of ≥1.0 kg in a week were defined as responders. The number and percentage of responders was calculated. Assessment of the response in Treatment Period-II was calculated by evaluating the changes in body weight between days 7 and 14. Urine samples were collected, to determine cumulative daily urine volume on days 1, 7, 8 and 14.
Safety evaluation
Safety assessment was performed throughout the study after the first administration of tolvaptan. The safety evaluation was based on the assessment of physical signs and symptoms, laboratory tests (haematology, clinical chemistry and urinalysis), vital signs (blood pressure, pulse rate and body temperature), and 12-lead electrocardiography. Blood samples to determine serum sodium concentrations were collected at baseline, at 4–8 h and at 22–24 h after dosing on days 1 and 8 (although they were not collected for patients on 7.5 mg/day, on day 8); and at 22–24 h after dosing on days 7 and 14. Blood samples were collected in sterile tubes containing heparin sodium (39 IU in 2 ml).
Statistical analyses
Analyses were performed on the full analysis set, which included all patients who received the trial drug at least once. Missing data at the final evaluation day were imputed by the last data obtained after the start of investigation (the ‘last observation carried forward’ method). Patients who received the trial drug at least once were included in the safety assessment.
Continuous data were presented as mean ± SD. Descriptive statistics in changes in body weight, abdominal circumference, serum sodium concentration and daily urine volume at each timepoint from baseline, and number of responders, were calculated. These analyses were conducted using the paired t-test. All statistical analyses were performed using SAS® software, version 9.1.3 (SAS Institute, Cary, NC, USA). A two-tailed P-value < 0.05 was considered statistically significant.
Results
A total of 71 patients were enrolled in this study, of whom 61 met the inclusion criteria. Figure 2 shows the patient disposition. Ten patients were withdrawn during the pretreatment observation period (ineligible n = 3, protocol violation n = 1, investigator’s decision n = 6). Fifty-one patients proceeded to the treatment period, of whom five were withdrawn due to adverse events (one each of malaise, increased serum potassium level, hepatic encephalopathy, obstructive hernia, decreased blood pressure) during Treatment Period-I. Three patients stopped at the end of Treatment Period-I because of improvement in hepatic oedema and did not advance to Treatment Period-II. Thirty patients continued to receive tolvaptan at 7.5 mg/day (defined as ‘7.5 mg Patients’) and 13 received the increased dose of tolvaptan at 15 mg/day (defined as ‘15 mg Patients’). Four patients continued to receive 7.5 mg/day in Treatment Period-II, despite not achieving a reduction of body weight ≥1.0 kg: three due to adverse events (insomnia, somnolent, serum creatinine increased) and one by the investigator’s decision. Two patients from each dose discontinued tolvaptan in Treatment Period-II: one due to adverse events at each dose, one patient on 7.5 mg at his own request and one on 15 mg due to insufficient efficacy (Figure 2). Patient demographics and baseline characteristics are shown in Table 1.
Flow chart indicating the recruitment and treatment of patients with hepatic oedema-associated ascites refractory to conventional diuretic treatment, included in a study to evaluate efficacy and safety of 7.5 mg/day tolvaptan with a dose increase to 15 mg/day in those with insufficient response at day 7. Baseline demographic and clinical characteristics of patients with hepatic oedema-associated ascites refractory to conventional diuretic treatment, included in a study to evaluate efficacy and safety of 7.5 mg/day tolvaptan with a dose increase to 15 mg/day in those with insufficient response at day 7 (n = 51). Data presented as mean ± SD or n (%) patients. Total bilirubin, serum albumin, prothrombin time-international normalized ratio, ascites and hepatic encephalopathy are scored 1–3, with 3 indicating most severe derangement. Chronic liver disease is classified into Child–Pugh class A (5–6), B (7–9) and C (10–15), employing a total score of the above-mentioned five clinical measures.
29
Patients with hepatic encephalopathy of Inuyama classification grade
23
≥II were excluded from the study.

The mean change in body weight from baseline in all patients (n = 46) who completed Treatment Period-I was significantly reduced by day 7 (P < 0.0001; Figure 3). Those who responded to 7.5 mg/day tolvaptan in Treatment Period-I (7.5 mg Patients) continued to respond in Treatment Period-II, with further significant mean decreases in body weight (P < 0.0001; Figure 4). Among 15 mg Patients, there were no mean decreases in in body weight on days 7 or 14.
Mean change from baseline in body weight in patients with hepatic oedema-associated ascites refractory to conventional diuretic treatment, treated with 7.5 mg/day tolvaptan for 7 days (Treatment Period-I). Numerical values shown on days 1 and 7 represent mean change from baseline in body weight ± SD (kg). *P < 0.0001 versus baseline; paired t-test. Mean change from baseline in body weight in patients with hepatic oedema-associated ascites refractory to conventional diuretic treatment, treated with 7.5 mg/day tolvaptan for 7 days (Treatment Period-I) and, in Treatment Period-II, with either 7.5 mg/day tolvaptan (circles) or 15 mg/day tolvaptan (triangles). Values represent mean change from baseline in body weight ± SD (kg). *P < 0.0001 versus baseline; paired t-test.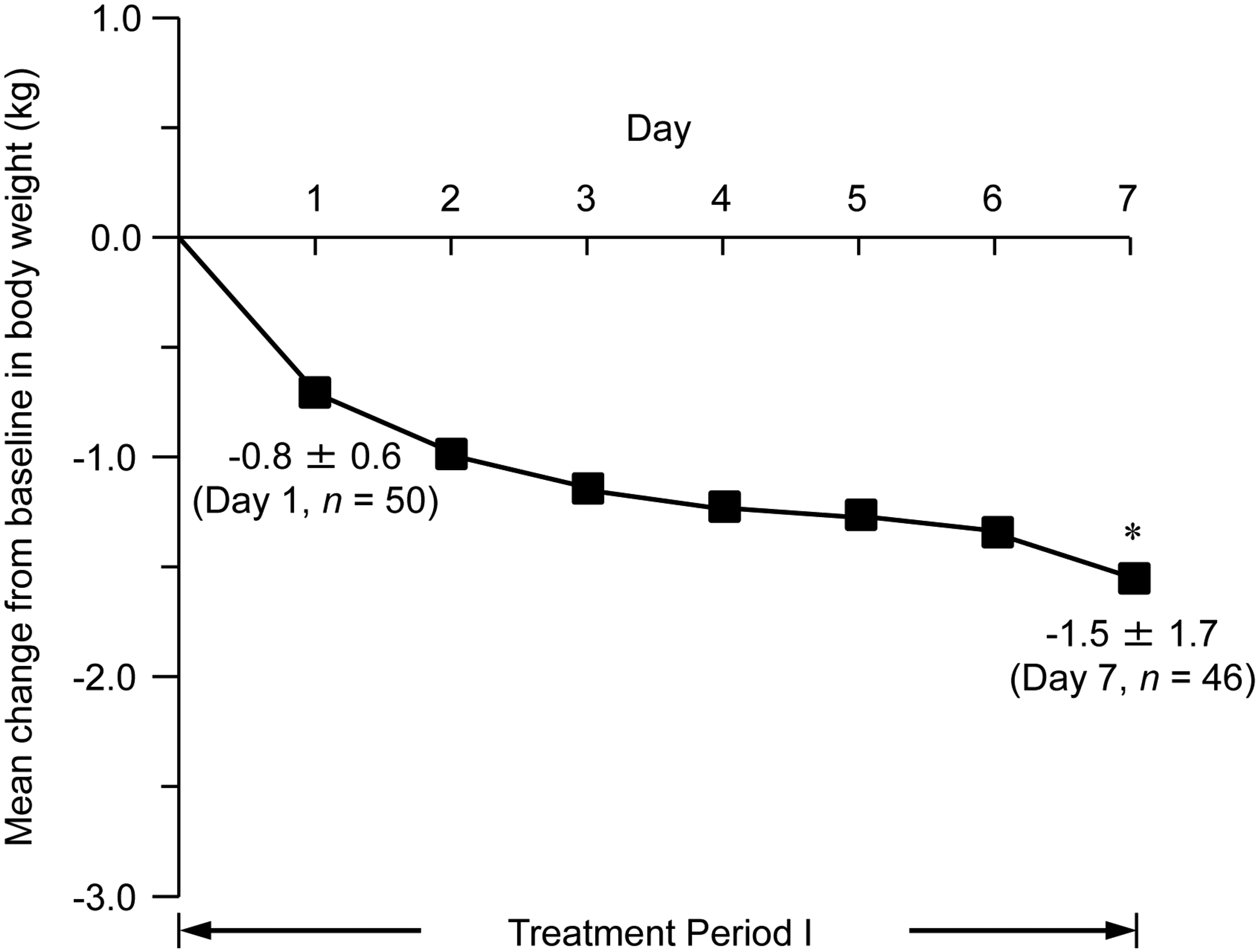

Rates of responders (patients with a body-weight reduction of ≥1 kg) after 7 days’ treatment with tolvaptan 7.5 mg (Treatment Period-I) and after an additional 7 days’ treatment with either 7.5 or 15 mg/day tolvaptan. Patients had hepatic oedema-associated ascites refractory to conventional diuretic treatment.

Data presented as n (%) patients.
Evaluation in Treatment Period-II was calculated by differences in body weight from day 7.
Patients who received 7.5 mg/day tolvaptan in Treatment Period-II achieved a body-weight reduction ≥1.0 kg in Treatment Period-I.
Patients who received 15 mg/day tolvaptan in Treatment Period-II had not achieved a body-weight reduction ≥1.0 kg in Treatment Period-I.
Four patients received 7.5 mg/day in Treatment Period-II despite, not achieving a body-weight reduction of ≥1.0 kg: three due to adverse events (insomnia, somnolent, serum creatinine increased) and one by the investigator’s decision.
Abdominal circumference at baseline and after 7 days’ treatment with tolvaptan 7.5 mg (Treatment Period-I) and after an additional 7 days’ treatment with either 7.5 or 15 mg/day tolvaptan, in patients with hepatic oedema-associated ascites refractory to conventional diuretic treatment.

Data presented as mean ± SD.
Patients who received 7.5 mg/day tolvaptan in Treatment Period-II had achieved a body-weight reduction ≥1.0 kg in Treatment Period-I.
Patients who received 15 mg/day tolvaptan in Treatment Period-II had not achieved a body-weight reduction ≥1.0 kg in Treatment Period-I.
Four patients received 7.5 mg/day in Treatment Period-II despite not achieving a body-weight reduction ≥1.0 kg: three due to adverse events (insomnia, somnolent, serum creatinine increased) and one by the investigator’s decision.
P < 0.0001 versus baseline; paired t-test.
Urine volume at baseline and during the first 7 days’ treatment with tolvaptan 7.5 mg (Treatment Period-I) and an additional 7 days’ treatment with either 7.5 or 15 mg/day tolvaptan in patients with hepatic oedema-associated ascites refractory to conventional diuretic treatment.

Data presented as mean ± SD.
Patients who received 7.5 mg/day tolvaptan in Treatment Period-II had achieved a body-weight reduction ≥1.0 kg in Treatment Period-I.
Patients who received 15 mg/day tolvaptan in Treatment Period-II had not achieved a body-weight reduction ≥1.0 kg in Treatment Period-I.
P < 0.05 versus baseline; paired t-test.
Serum sodium concentrations at baseline and during the first 7 days’ treatment with tolvaptan 7.5 mg (Treatment Period-I) and an additional 7 days’ treatment with either 7.5 or 15 mg/day tolvaptan, in patients with hepatic oedema-associated ascites refractory to conventional diuretic treatment.

Data presented as mean ± SD.
Patients who received 7.5 mg/day tolvaptan in Treatment Period-II had achieved a body-weight reduction ≥1.0 kg in Treatment Period-I; blood samples were not collected from 7.5 mg Patients on day 8, therefore, data on serum sodium concentration are not available.
Patients who received 15 mg/day tolvaptan in Treatment Period-II had not achieved a body-weight reduction ≥1.0 kg in Treatment Period-I.
P = 0.00, day 7 versus baseline; dP = 0.033, day 7 versus baseline; eP = 0.009, day 14 versus baseline; paired t-test.
Total adverse events and adverse events with an incidence of ≥5% reported during the first 7 days’ treatment with tolvaptan 7.5 mg (Treatment Period-I) and an additional 7 days’ treatment with either 7.5 or 15 mg/day tolvaptan, in patients with hepatic oedema-associated ascites refractory to conventional diuretic treatment (n = 51).
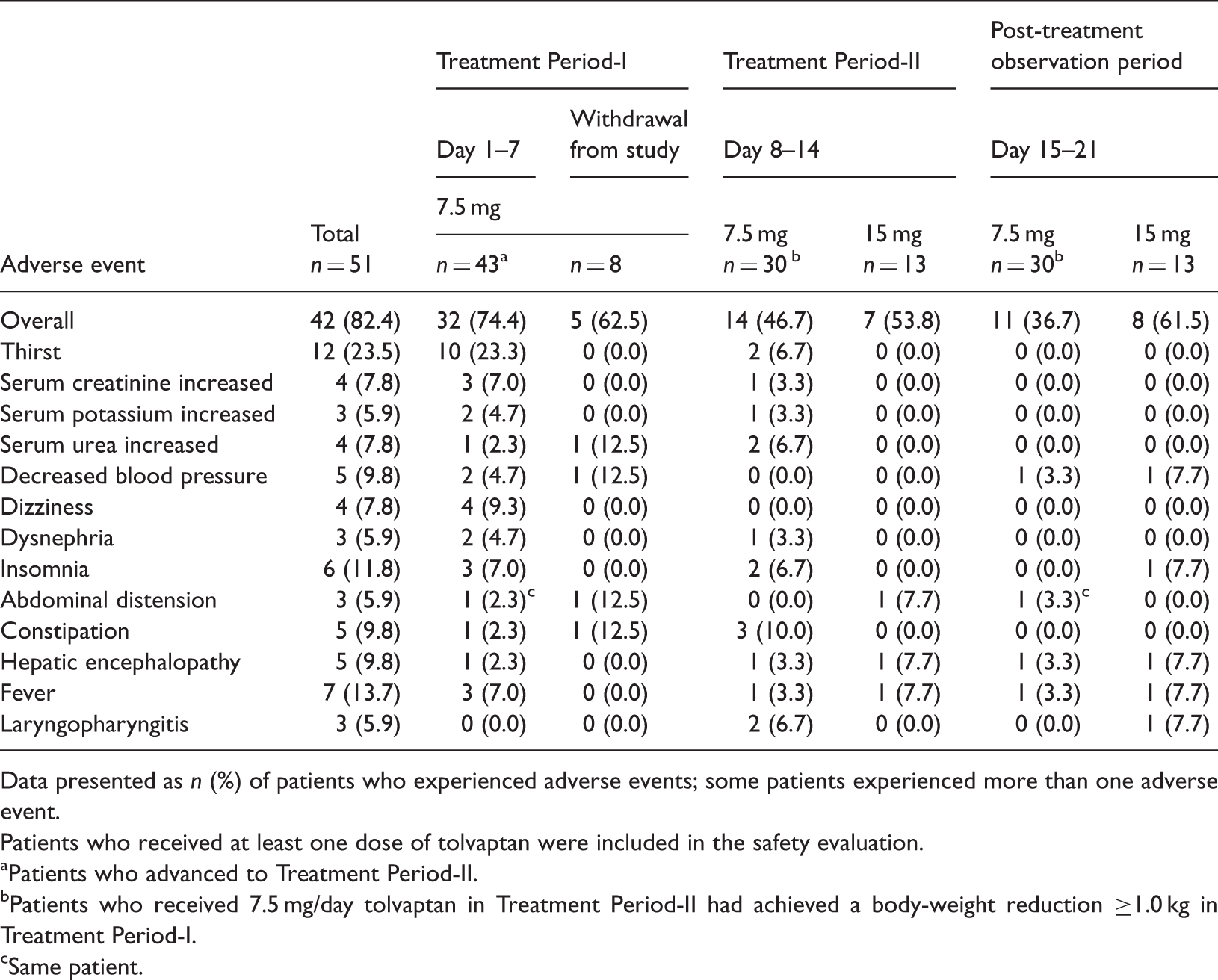
Data presented as n (%) of patients who experienced adverse events; some patients experienced more than one adverse event.
Patients who received at least one dose of tolvaptan were included in the safety evaluation.
Patients who advanced to Treatment Period-II.
Patients who received 7.5 mg/day tolvaptan in Treatment Period-II had achieved a body-weight reduction ≥1.0 kg in Treatment Period-I.
Same patient.
Discussion
The present study demonstrated that just over half of the enrolled patients with hepatic oedema who had failed to respond to conventional diuretic treatment responded to 7 days’ treatment with tolvaptan 7.5 mg/day. Those patients who responded to this treatment continued to achieve body weight decreases on the same dose of tolvaptan during the subsequent 7 days. In those who did not initially respond to 7.5 mg/day tolvaptan, a dose increase to 15 mg/day meant that three additional patients responded. Additionally, ascites improved in 7.5 mg Patients, as shown by continuous decreases in abdominal circumference.
A loop diuretic, such as furosemide, has superior diuretic action to other types of diuretics, although it is associated with adverse drug reactions including hyponatraemia and renal impairment.3,4,27,28 Combination therapy with tolvaptan plus a loop diuretic in the present study led to a sustained decrease in body weight during the entire treatment period, in patients who were responders in Treatment Period-I, with minimum effect on serum sodium concentration. Thus, the combination therapy of the aquaretic tolvaptan with a loop diuretic could be a novel treatment option in patients with hepatic oedema without heightened risk of electrolyte imbalance. 14 The use of tolvaptan presupposes concomitant use with other conventional diuretics in Japan. Tolvaptan increases serum sodium concentrations as a result of improvement in hyponatraemia. Although conventional diuretics, particularly loop diuretics with urinary sodium excretion, do not exert adequate diuresis in patients with hyponatraemia, tolvaptan is expected to exert its sufficient diuresis by increasing the serum sodium concentration. Thus, even when patients do not respond to conventional diuretics alone, diuretics should be administered concomitantly with tolvaptan.
The safety analysis showed that adverse events were observed in 82% of patients during the 14-day treatment period. The most commonly observed adverse event was thirst, as predicted from the pharmacological action of tolvaptan. Safety analyses suggested that the incidence of thirst was reduced over time, as a lower incidence was observed in the second 7-day period.
There were some limitations to the present study. A treatment period >14 days and a higher dose than 15 mg/day tolvaptan could be allocated to patients who do not respond to 7.5 mg/day. 22 This single-arm study was designed to evaluate efficacy and safety of tolvaptan at 7.5 mg/day for 14 days. For this purpose, the sample size was sufficient.
In conclusion, based on the current study, tolvaptan is considered to be an effective aquaretic agent, offering a novel treatment option for hepatic oedema, in patients with insufficient response to conventional diuretics.
Declaration of conflicting interest
Dr Isao Sakaida received research grants from Otsuka Pharmaceutical (Tokyo, Japan), MSD (Tokyo, Japan), Chugai Pharmaceutical (Tokyo, Japan) and Takeda Pharmaceutical (Osaka, Japan), and served as a consultant for Otsuka Pharmaceutical with payment of a consultancy fee to Yamaguchi University Graduate School of Medicine (Ube, Japan). Mitsuru Okada is an employee of Otsuka Pharmaceutical. No other author has any conflict of interest to declare in relation to this article.
Footnotes
Funding
This study was funded by Otsuka Pharmaceutical Company, Tokyo, Japan.
Acknowledgements
In addition to the authors, the following investigators participated in the ASCITES 14-Day Administration STUDY GROUP: Eiji Kajiwara (Steel Memorial Yawata Hospital, Kitakyushu), Go Kobayashi (Sendai Open Hospital, Sendai), Hideyuki Nomura (SHIN-KOKURA Hospital, Kitakyushu), Hirayuki Enomoto (Hyogo College of Medicine Hospital, Nishinomiya), Hiroshi Mieno (Hiroshima General Hospital of West Japan Railway Company, Hiroshima), Hiroyuki Kokuryu (Kyoto-Katsura Hospital, Kyoto), Junichi Sugihara (Gifu Prefectural General Medical Center, Gifu), Kazuhiro Katayama (Osaka Medical Center for Cancer and Cardiovascular Diseases, Osaka), Kazuyoshi Matsumura (Shizuoka General Hospital, Shizuoka), Kenji Miyakoda (Kurume University Hospital, Kurume), Kunihiko Shimatani (National Hospital Organization Hiroshima-Nishi Medical Center, Otake), Masafumi Kumamoto (Kurume University Hospital, Kurume), Masahiro Araki (Ibaraki Prefectural Central Hospital, Kasama), Masahito Uemura (Nara Medical University Hospital, Kashihara), Masashi Yoneda (Aichi Medical University Hospital, Aichi-gun), Motoyuki Yoshida (Nara Medical University Hospital, Kashihara), Nobuyoshi Tanaka (FukuiKen Saiseikai Hospital, Fukui), Ryujin Endo (Iwate Medical University Hospital, Morioka), Satoru Kakizaki (Gunma University Hospital, Maebashi), Satoru Kaneda (National Hospital Organization Chiba Medical Center, Chiba), Takahisa Kayahara (Shizuoka General Hospital, Shizuoka), Takashi Kumada (Ogaki Municipal Hospital, Ogaki), Toru Ishikawa (Saiseikai Niigata Daini Hospital, Niigata), Toshihiko Nouchi (Showa General Hospital, Kodaira), Toshiyuki Otsuka (National Hospital Organization Nishigunma Hospital, Shibukawa), Toyokazu Fukunaga (Kitano Hospital, The Tazuke Kofukai Medical Research Institute, Osaka), Tsukasa Aihara (Meiwa Hospital, Nishinomiya), Yoshiro Baba (Kagoshima Kouseiren Hospital, Kagoshima), Yutaka Sasaki (Kumamoto University Hospital, Kumamoto).