
Abstract
Objective
The function of Ras-related C3 botulinum toxin substrate1 (Rac1) in the progression of cervical cancer is unclear. This study used RNA interference technology to explore the involvement of Rac1 in the regulation of cervical cancer cells.
Methods
A short hairpin (sh) RNA plasmid targeting Rac1 was constructed and transfected into HeLa cells. Rac1 mRNA and protein levels were investigated by reverse transcription–polymerase chain reaction and Western blot, respectively. Cell proliferation and cisplatin chemosensitivity were determined using the methyl thiazolyl tetrazolium assay. The Matrigel™ assay and flow cytometry were used to assess cell invasion and apoptosis, respectively. The concentration of matrix metalloproteinase (MMP)-2 in cell supernatants was detected by enzyme-linked immunosorbent assay.
Results
Rac1 expression was significantly downregulated at the mRNA and protein levels in HeLa cells transfected with Rac1 shRNA, and the cell proliferation and invasion capability of cells was decreased. Rac1 downregulation was associated with a decrease in MMP-2 secretion, and increased cell chemosensitivity to cisplatin and cisplatin-induced apoptosis.
Conclusions
Rac1 may play an important role in cervical cancer progression and could be a potential target for anticancer therapy.
Keywords
Introduction
Cervical cancer is the second most common cancer in women worldwide, with ∼55 000 new cases and ∼310 000 deaths annually, ∼80% of which occur in developing countries. 1 Due to effective implementation of cytological screening and treatment protocols, the incidence and mortality rates for cervical cancer in developed countries have both declined dramatically over the past two decades. Disease incidence, however, remains high in regions where there is a lack of resource for cervical cancer screening, such as South America, Africa and South-East Asia. 2 In particular, nearly one-third of patients will die from recurrent, metastatic or pharmacoresistant forms of cervical cancer. 3 Thus, a better understanding of the molecular mechanisms underlying proliferation, invasion and survival in cervical cancer is critical, in order to facilitate the development of appropriate therapeutic strategies.
Ras-related C3 botulinum toxin substrate 1 (Rac1) is highly expressed in cervical premalignant lesions and cervical cancer cells, which suggests that it may play an important role in cervical cancer progression. 4
The RAC1 gene encodes a 21-kDa GTP-binding protein belonging to the Rho family of small GTPases, which regulate cellular function during embryonic development and tumour invasion. 5 Rac1 is ubiquitously expressed in eukaryotic cells, regulates formation of lamellipodia, and plays an important role in tumourigenesis, invasion and metastasis. 6 Rac1 is overexpressed in many malignant tumours including testicular cancer, gastric cancer, hepatocellular carcinoma, leukaemia and oral squamous cell carcinoma.7–11 Depletion of Rac1 by small-interfering RNA (siRNA) may strongly inhibit lamellipodia formation and cell invasion in glioblastoma and breast cancer cells. 12 In addition to controlling the actin cytoskeleton, Rac1 regulates cell invasion by stimulating signalling pathways. It has been reported that Rac1 controls the expression and activity of different matrix metalloproteinases (MMPs) such as MMP-1, MMP-2 and MMP-14, to remodel the extracellular matrix (ECM). 13 The MMPs belong to a family of proteases, among which MMP-2 and MMP-9 in particular are known to play crucial roles in tumour invasion, angiogenesis and subsequent distant metastasis by proteolytic degradation of ECM. 14 Mendoza-Catalán et al. 4 have shown that Rac1 is overexpressed in cervical premalignant lesions and cervical cancer cells, but no further research into the mechanism by which Rac1 affects cervical cancer has been published.
In order to explore the role of Rac1 in cervical cancer progression, the present study used RNA interference (RNAi) technology to knock down Rac1 expression in HeLa cells and then determine the effects on cellular proliferation, invasion, apoptosis and chemosensitivity to cisplatin. It was hoped that the findings may provide experimental evidence for further molecular mechanisms in relation to the malignant progression of cervical cancer and adjuvant chemotherapeutic strategies for this disease.
Materials and methods
Cell culture and reagents
HeLa cells, a human cervical epithelial adenocarcinoma cell line (obtained from the Scientific Research Foundation of the Second Affiliated Hospital of Harbin Medical University, Harbin, China) were cultured in Dulbecco's Modified Eagle’s Medium (DMEM, Gibco BRL, Life Technologies, Gaithersburg, MD, USA), supplemented with 10% fetal bovine serum (FBS) in the absence of antibiotics at 37 ℃ in a humidified atmosphere, containing 5% CO2. Cisplatin was purchased from the Hansoh Pharmaceutical Group (JiangSu, China).
Rac1 shRNA and transient transfection procedure
A commercially-available vector, pGPU6/GFP/Neo (Shanghai Sangon Biotech Co, Shanghai, China), was used to generate short hairpin (sh) RNA specific for Rac1. The human RAC1 gene-coding sequence was obtained from GenBank® (http://www.ncbi.nlm.nih.gov/genbank/) where four sets of shRNA sequence targeting the human Rac1 gene are listed: Rac1-sh294 (5′-GCCTACTGATCAGTTACACAA-3′); Rac1-sh320 (5′-GCATTTCC TGGAGAATATATC-3′); Rac1-sh365 (5′-GCCAATGTTATGGTAGATGGA-3′); Rac1-sh394 (5′-GAATCTGGGCTTATGGGATAC-3′). A nonspecific shRNA was designed as a negative control (Rac1-shNC) (5′-GTTCTCCGAACGTGTCACGT-3′). These oligonucleotides were synthesized and subcloned into the BamHI and PstI restriction sites of the vector at 22℃ for 1 h. HeLa cells were plated in six-well plates at a concentration of 6 × 104 cells per well, and incubated overnight. Cells were then transfected with Rac1-shRNAs (2 µg plasmid in 250 µl DMEM) using 5 µl Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Untransfected HeLa cells were included as a blank control group. The success of the transfection was determined 48 h later by inverted fluorescence and phase-contrast microscopy (LEICA DMIL-PH1, Leica Microsystems, Wetzlar, Germany) using three randomly-selected fields of vision.
RT–PCR analysis of Rac1 mRNA
Reverse transcription–polymerase chain reaction (RT–PCR) analysis was performed as follows. Total RNA was extracted from HeLa cells (1 × 107 cells per well) using TRIzol® (Invitrogen). The RT reaction was performed with 1 µg total RNA in 20 ‐µl reactions using the ImProm-II™ Reverse Transcription System (Promega, Madison, WI, USA), incubated at 42 ℃ for 15 min and heated at 94 ℃ for 5 min. The primer sequences for Rac1 were 5′-GCGGCACCACTGTCCCAACA-3′ (forward) and 5′-AGCGCCGAGCACTCCAGGTA-3′ (reverse), and for β-actin were 5′-AGGTCGGAGTCAACGGATTTGGTCG-3′ (sense) and 5′-TGGCCAGGGG TGCTAAGCAGT-3′ (antisense). PCR was performed using a Roche LightCycler® (Bio-Rad, Hercules, CA, USA) under the following conditions: 30 amplification cycles of denaturation at 94 ℃ for 30 s, annealing at 58 ℃ for 30 s and extension at 72 ℃ for 50 s, followed by a final extension at 72 ℃ for 10 min. PCR products were subjected to electrophoresis on 1% agarose gel and visualized by ethidium bromide staining. Relative band intensities were detected using ImageJ software (available at http://rsb.info.nih.gov/ij/). The density of each band was normalized against that of β-actin.
Western Blot analysis of Rac1 protein
Cells (1 × 107 cells per well) were lysed by precooled lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM phenylmethylsulphonyl fluoride, 1 mM ethylenediaminetetra-acetic acid, 1% NP-40, 1% sodium deoxycholate and 0.1% sodium docecyl sulphate [SDS], pH 7.4) and the protein content of the lysates was assessed using a BCA Protein Assay Kit (Beyotime Institute of Biotechnology, Shanghai, China). Equal amounts of cell lysate (20 µg) were separated by 10% SDS polyacrylamide gel electrophoresis and were electroblotted onto polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). Blotted membranes were blocked with 5% skimmed milk in Tris-buffered saline containing Tween (TBST, pH 7.6) at room temperature for 1 h. Blots were then incubated overnight at 4 ℃ with a 1 : 400 dilution of antihuman Rac1 mouse monoclonal antibody followed by incubation for 1 h at room temperature with a 1 : 1000 dilution of horseradish peroxidase-conjugated antimouse goat monoclonal antibody (both from Santa Cruz Biotechnology, Santa Cruz, CA, USA). After each incubation, membranes were washed three times each for 15 min using 0.05% TBST. Protein bands were visualized using a diaminobenzidine colouration kit and electrochemiluminescence analysis system (Wuhan Boster Biological Technology, Wuhan, China) according to the manufacturer’s instructions.
MTT assay for assessment of cell proliferation and chemosensitivity to cisplatin
Exponentially growing HeLa cells were seeded into 96-well microtitre plates at a concentration of 5 × 10 4 cells per well, and allowed to attach overnight before Rac1 shRNA transfection. Following 24, 48, 72 and 96 h of culture, cells were incubated with 20 µl of MTT (5 mg/ml; Sigma-Aldrich, St Louis, MO, USA) at 37 ℃ for 4 h after which the medium was decanted and the reaction stopped with 150 µl of dimethyl sulphoxide (Sigma-Aldrich). The spectrometric absorbance of each sample was measured at 490 nm by an automatic microplate reader (Bio-Rad).
For assessment of chemosensitivity, exponentially growing cells were seeded into 96-well microtitre plates at a density of 5 × 104 cells per well, and left overnight at 37° C. Two days after transfection, the cells were treated with 0 – 40 µg/ml cisplatin for 24 h before incubation with MTT, as described above. The proliferation inhibition ratio was calculated from the spectrometric absorbance of each cisplatin group.
Matrigel™ invasion assay
Transwell® chambers with 8 -µm pore size polycarbonate membranes (Costar®, both from Corning, Lowell, MA, USA) were coated with 60 µl Matrigel™ (Becton, Dickinson and Co., Franklin Lakes, NJ, USA) and incubated at 37 ℃ for 30 min. A 200 -µl suspension of 5 × 104 cells was seeded onto the upper chamber and cultured with serum-free DMEM medium for a further 24 h at 37 ℃. Lower chambers were filled with 600 µl DMEM /10% FBS. The Matrigel™ was scraped away and the remaining polycarbonate membranes were stained with 0.1% crystal violet for 10 min. The number of cells invading the Matrigel™ and attaching to the lower surface of the membranes were counted by light microscopy (Olympus BX40, Olympus Optical Co., Tokyo, Japan).
ELISA for detection of MMP-2
Cells were incubated for 48 h after transfection and the culture supernatant was collected by centrifugation at 3000 rpm for 15 min at room temperature (Centrifuge L-600, Shanghai Lixinjian Centrifuge, Shanghai, China). The concentration of MMP-2 in the culture supernatant was detected by an enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN, USA), according to the manufacturer's instructions. Optical density was measured by an ELISA reader (KC100, Caretium Medical Instruments, Shenzhen, China) at 450 nm.
Flow cytometry analysis of cisplatin-treated cells
Measurement of cell apoptosis assessment was performed by flow cytometry 48 h after transfection with Rac1-shRNA. Transfected cells were treated with 10 µg/ml cisplatin for 24 h followed by incubation with 5 µl annexin V–fluoresceine-isothiocyanate (20 µg/ml) and 5 µl proprium iodide (50 µg/ml) (Sigma-Aldrich) for 15 min at 37 ℃. Cell apoptosis was measured by a Cytomics FC 500 MPL flow cytometer (Beckman Coulter, Brea, CA, USA). CXP software, version 2.2 (Beckman Coulter) was used to analyse the data.
Statistical analyses
Each experiment was performed in triplicate. Statistical analyses were carried out using SPSS® statistical software, version 19.0 (SPSS, Inc. Chicago, IL, USA) for Windows®. Data were expressed as mean ± SD. Analysis of variance (ANOVA) experiments (one-way ANOVA, factorial ANOVA and repeated-measures ANOVA), followed by the Student–Newman–Keuls test were performed, with P < 0.05 considered statistically significant.
Results
The success of transfection of HeLa cells with Rac1 shRNA was determined by fluorescence microscopy, which demonstrated that >65% of cells emitted green fluorescence 48 h after transfection (Figure 1A). RT–PCR analysis showed that at 48 h after transfection, Rac1 was effectively knocked down by sh294, but that sh365, sh320 and sh394 had no obvious effect (Figures 1B and C). Western blot analysis demonstrated that all four shRNA sequences effectively knocked down Rac1, with sh294 having the highest suppression efficacy (Figures 1D and E). These results confirmed that sh294 effectively interfered with Rac1 production; the effect of Rac1 downregulation on cell proliferation and invasion was, therefore, determined in HeLa cells transfected with the Rac1-sh294 sequence.
Detection of Rac1-short hairpin (sh) RNA transfection efficiency and interference efficiencies. (A) (a) Representative fluorescence micrograph of HeLa cells transfected with Rac1-sh294 containing green fluorescent protein (positive cells emitted green fluorescence); (b) Inverted phase-contrast micrograph of HeLa cells transfected with Rac1-sh294 (×200). (B) Representative reverse transcription–polymerase chain reaction analysis (RT–PCR) of Rac1 mRNA extracted from cervical cancer HeLa cells, 48 h after transfection with shRNA sequences specific for Rac1. (C) Quantitative analysis of the relative mean ± SD Rac1 mRNA levels as determined by RT–PCR (n = 3). (D) Representative Western blot analysis of Rac1 protein expression in cervical cancer HeLa cells 48 h after transfection with shRNA sequences specific for Rac1. (E) Quantitative analysis of the relative mean ± SD Rac1 protein levels as determined by Western blot (n = 3). *P < 0.05 (analysis of variance procedures, followed by Student–Newman–Keuls test). Untransfected HeLa cells used as blank controls; β-actin used as an internal control; DL2000 DNA marker was used; bp, base pairs; NC, negative control.
Compared with untransfected HeLa cells (blank control group), cell proliferation viability significantly decreased in cells transfected with Rac1 sh294 (Figure 2); the proliferation inhibition ratios were 40.85% ± 0.013, 35.38% ± 0.010 and 29.47% ± 0.021 at 48 h, 72 h and 96 h, respectively (P < 0.05, at each timepoint). There were no significant differences in cell proliferation between the control groups (blank controls and shNC transfected cells).
Effect of Rac1 downregulation on cell proliferation, as determined by methyl thiazolyl tetrazolium assay. The growth curve showed that the cell proliferation viability of HeLa cells transfected with Rac1-short hairpin (sh) RNA-294 decreased compared with untransfected cells (blank control) at all timepoints. *P < 0.05 (analysis of variance procedures, followed by Student–Newman–Keuls test). NC, negative control.
The numbers of cells penetrating Matrigel™ and adhering to the membrane in the blank control, shNC and sh294 groups were 47.0 ± 4.58, 44.0 ± 3.61, 16.67 ± 1.53, respectively (Figure 3A). A decrease in cell numbers in the sh294 group was observed, compared with the control groups (P < 0.05). Both control groups had similar ability to penetrate the membranes; with no significant differences being observed.
Effect of Rac1 downregulation on cell invasion and chemosensitivity to cisplatin. (A) Representative light micrographs (×200) demonstrating the cell invasion ability of HeLa cells 48 h after transfection with Rac1-short hairpin (sh) RNA-294 using the Matrigel™ invasion assay. (B) Reduction in the secretion of matrix metalloproteinase (MMP)-2 following Rac1 knockdown as determined by enzyme-linked immunosorbent assay of in HeLa cell supernatants 48 h post-transfection. (C) Effect of Rac1 downregulation on the chemosensitivity of HeLa cells to cisplatin as determined by methyl thiazolyl tetrazolium assay 48 h post-ransfection. Data are shown as mean ± SD and cell viability was calculated as AR490 (experimental group)/AR490 (negative control) (n = 3). *P < 0.05 (Analysis of variance procedures, followed by Student–Newman–Keuls test). Untransfected HeLa cells used as blank controls. NC, negative control.
Using ELISA, it was demonstrated that the concentration of MMP-2 in the supernatants of HeLa cells transfected with Rac1-sh294 decreased significantly compared with the two control groups 48 h after transfection (P < 0.05; Figure 3B). No significant differences were observed between the blank control and shNC transfected cells.
Cell survival decreased with increasing concentrations of cisplatin across all treatment groups (blank controls, negative controls and cells transfected with Rac1-sh294) (Figure 3C). These decreases were significantly greater in the sh294 group at all cisplatin concentrations, compared with blank controls and negative controls (P < 0.05).
Flow cytometry showed that after treatment with 10 µg/ml cisplatin or cisplatin plus shNC transfection, the early apoptosis rate increased to 15.0% and 15.5%, respectively, compared with the blank control (P < 0.05); no significant differences were observed between the two cisplatin groups (Figure 4A). Following treatment with cisplatin plus Rac1-sh294 transfection, the early apoptosis rate rose to 23.6%. This difference was statistically significant compared with the other three treatment groups (P < 0.05; Figure 4B).
Effect of Rac1 downregulation on cell apoptosis. (A) Flow cytometric analysis of cell apoptosis using Annexin V–fluoresceine-isothiocyanate (FITC)/proprium iodide staining in HeLa cells treated with cisplatin, cisplatin plus transfection with Rac1-short hairpin (sh) negative control (NC) RNA or cisplatin plus transfection with Rac1-sh294. (B) Quantitative analysis of the mean ± SD percentage of cells in the early stage of apoptosis. Cells were treated with 10 µg/ml cisplatin 24 h before transfection. *P < 0.05 (analysis of variance procedures, followed by Student–Newman–Keuls test).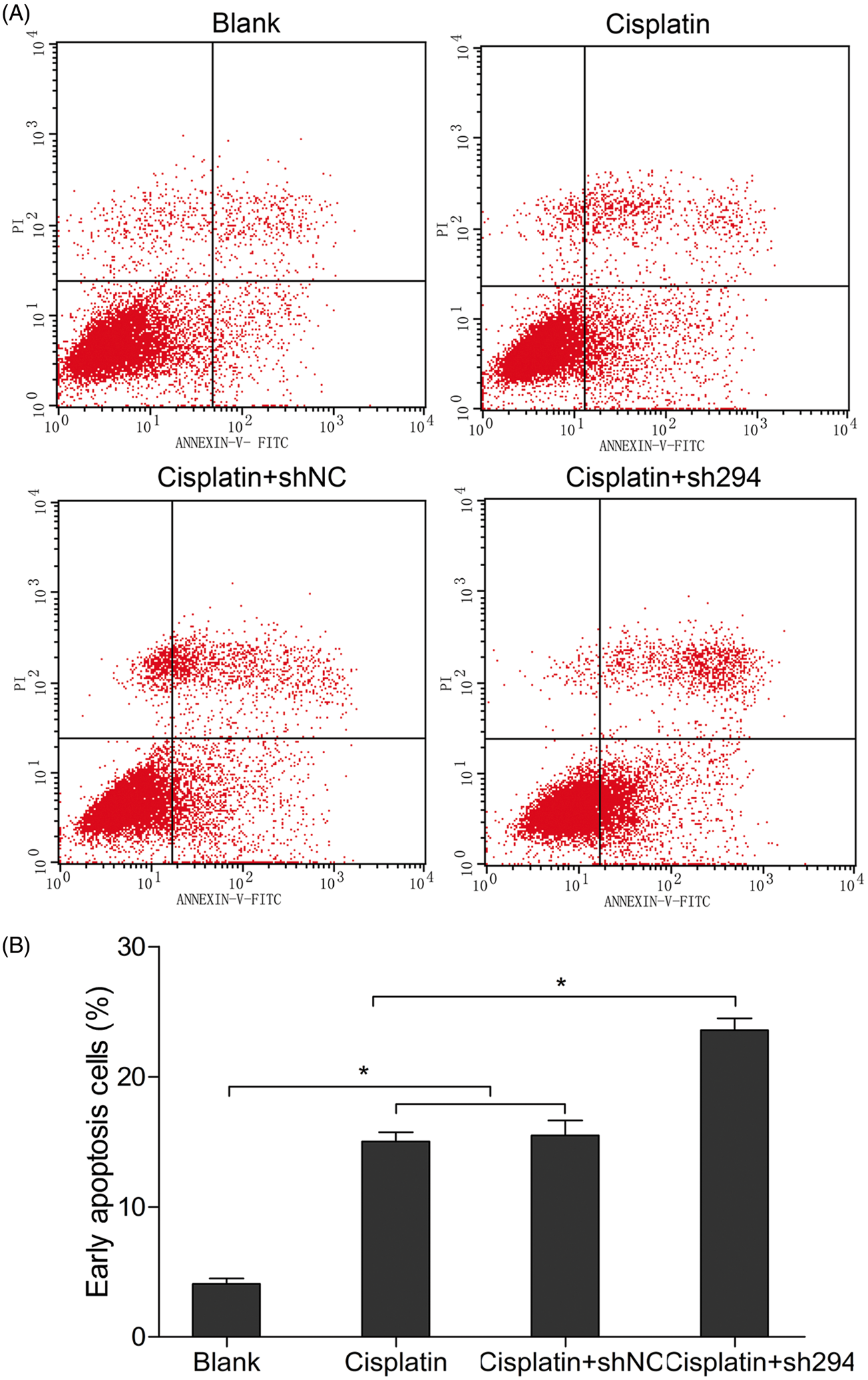
Discussion
The Rho small GTPases family includes Rho, Rac and Cdc42 proteins, which act as molecular switches and control signalling pathways that regulate many cellular functions (such as cell morphology, gene expression, and cell proliferation and survival). 15 Rac1 may also mediate distinct actin cytoskeleton changes and produce lamellipodia at the cell border. 15 These cellular functions are important in tumourigenesis and cancer progression. 16 Rac1 has been found to be overexpressed in various cancer types, and possesses multiple functions including promotion of pancreatic cancer-cell proliferation via activation of Rac1-dependent superoxide generation; 17 Rac1 also suppressed apoptosis of epithelial cells in suspension via phosphoinositide 3-kinase, contributing to cell malignant transformation and tumourigenesis. 18 One report suggested that Rac1 overexpression could mediate drug resistance via impaired trastuzumab-mediated endocytic downregulation of ErbB2 and enhanced extracellular signal-regulated kinase activity, in breast cancer cells. 19 Together, these findings indicate that Rac1 is a major mediator of malignant-cell behaviour, although its function in cervical cancer remains poorly understood.
RNA interference is a gene-silencing technique that works at the post-transcriptional level; shRNA mediates sequence-specific inhibition of gene expression. In the present study, we used RNAi technology to construct a Rac1-shRNA expression vector, followed by transfection into the cervical cancer cell line, HeLa. The findings indicated that mRNA and protein levels of Rac1 in HeLa cells post-transcription were significantly downregulated, compared with levels observed in the control groups. Results of MTT analyses revealed that proliferation of HeLa cells in which the Rac1 gene had been knocked down was decreased, which was in contrast to findings observed in the controls. The present data are consistent with other studies investigating the role of Rac1 in cell proliferation.20 – 22 Inhibition of RAC1 in rat aortic smooth-muscle cells was shown to reduce levels of intracellular reactive oxygen species and cellular proliferation induced by platelet-derived growth factor. 20 In another report, Rac1 activity was demonstrated to regulate proliferation of aggressive metastatic melanoma, in part, via its effect on NF-κB. 21 Research in prostate cancer also showed that signalling through a cSrc or a protein kinase C pathway regulated prostate cancer cell proliferation via RAC1, and that RAC1 inhibition suppressed cell proliferation in all prostate cancer cell lines studied. 22
Cell invasion experiments conducted as part of the present study indicated that the velocity of cells transfected with Rac1-sh294 to penetrate Matrigel™ was reduced, compared with the two control groups. Accordingly, the concentration of MMP-2 in cells supernatants from the Rac1-sh294 group decreased substantially. MMP-2 is one of the downstream target genes of Rac1 13 and the present study, therefore, suggests that Rac1 may promote invasion of cervical cancer, at least in part, by regulating MMP-2 expression. Tumour cell invasion and metastasis comprise two essential processes: migration and proteolysis. Migration is initiated by actin polymerization at the cell's leading edge and at the lamella extension in the direction of motion, both of which are regulated mainly by Rac1. 23 MMP-2 (gelatinase-A, type IV collagenase) also plays an important role in tumour invasion via proteolytic degradation of major components of the basement membrane, such as type IV collagen and laminin. 24 Other research has indicated that membrane-type-1 (MT1)-MMP could initiate the activation of MMP-2 in a process that could be inhibited by tissue inhibitor of metalloproteinase (TIMP)-2. Strongin et al. 25 proposed that MT1-MMP and TIMP-2 form a receptor complex of MMP-2, potentiating proteolysis of bound MMP-2 by adjacent active MT1-MMP. Rac1 activity facilitates a shift of the balance between MT1-MMP and TIMP-2 towards MT1-MMP expression, thereby increasing type I collagen-induced proteolytic activity of MMP-2. 13 Sustained activation or overexpression of Rac1 could promote invasion and metastasis of murine T-lymphoma cells and many epithelial cells.15,26,27 Other research has, however, indicated that activation of Rac1 promoted E-cadherin-mediated cell–cell adhesion and selectively upregulated TIMP-1 and TIMP-2, thereby inhibiting epithelial cell invasion. 28 These seemingly opposing effects of Rac1 on migration and invasion may be dependent on the cell substrate used and the cell type studied.
Historically, cisplatin is the most active drug against cervical cancer, with response rates of ∼40% in chemotherapy-naïve patients who have advanced/metastatic or recurrent disease. 29 Cisplatin is believed to mediate its cytotoxic properties through preferential binding to nuclear DNA and subsequent interference with normal transcription and/or DNA replication mechanisms, thus leading to apoptosis. 30 Increased repair activity of platinum-DNA adducts can contribute to cisplatin resistance, and inhibition of DNA repair can enhance cytotoxicity and induce apoptosis. Research on the effects of Rac1 on the modulation of apoptosis found that transient transfection of dominant-negative N17-Rac1 led to a decrease in DNA repair activity, 31 that Rac1 prevented cisplatin-induced apoptosis by suppressing p38 activation in NIH3T3 cells, 32 and that constitutively active Rac1 provided a survival signal under various stimuli in fibroblasts and melanoma cells.33,34 Results from the MTT assay in the present study revealed that downregulation of Rac1 increased chemosensitivity to cisplatin in HeLa cells. Flow cytometric analyses also showed that cisplatin-induced apoptosis was enhanced when Rac1 was inhibited. These results were consistent with previous reports,31–34 although other studies have suggested that activation of Rac1 GTPase is involved in apoptosis in fibroblasts and epithelial cells.35,36 Such discrepancies in the effects of Rac1 on apoptosis may be due to the different cell types studied, or due to the specificity of different extracellular stimuli. Further studies, to determine the function of Rac1 and the exact mechanisms involved in the development of cervical cancer in vivo, are warranted.
In conclusion, the present study showed that shRNA targeting Rac1 significantly downregulated Rac1 mRNA and protein in cervical cancer cells, and inhibited cell proliferation and invasive activity, which may, in part, be related to Rac1-mediated regulation of MMP-2 expression. The results also indicated that downregulation of Rac1 could increase the in vitro chemosensitivity of HeLa cells to cisplatin. Furthermore, the data suggest that Rac1 may play an important role in regulating cervical cancer cell apoptosis, and that cisplatin-induced apoptosis could be enhanced by Rac1 downregulation. Taken together, the findings presented here indicate that Rac 1 may be a potential target for antitumour drugs and provide new insights into the development of gene therapy technology for treating women with cervical cancer.
Footnotes
Acknowledgements
This work was supported by a grant from the Youth Foundation (QN2010-13) of the Second Affiliated Hospital of Harbin Medical University.
Declaration of conflicting interest
The authors have no conflicts of interest to declare in relation to this article.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.